
Abstract
We present first-principles studies of the optical absorbance of the group IV honeycomb crystals graphene, silicene, germanene, and tinene. We account for many-body effects on the optical properties by using the non-local hybrid functional HSE06. The optical absorption peaks are blueshifted due to quasiparticle corrections, while the influence on the low-frequency absorbance remains unchanged and reduces to a universal value related to the Sommerfeld fine structure constant. At the Dirac points spin–orbit interaction opens fundamental band gaps; parabolic bands with a very small effective mass emerge. Consequently, the low-frequency absorbance is modified with a spin–orbit-induced transparency region and an increase of the absorbance at the fundamental absorption edge.
Export citation and abstract BibTeX RIS
1. Introduction
Graphene, the flat two-dimensional (2D) honeycomb lattice of carbon atoms with π-bonding, has attracted much attention due to its unique properties [1]. It provides massless Dirac fermions in the low energy regime due to zero-energy gaps and a linear dispersion at the six corners K and K' of the hexagonal Brillouin zone (BZ). Besides graphene, the 2D honeycomb structure of silicon has also recently been discovered and named as silicene [2–4]. In contrast to graphene, the silicon atoms are buckled, leading to a partial sp3 hybridization of the electronic system. Nevertheless, the electronic properties of graphene and silicene are similar because of the same hexagonal symmetry. The prediction of linear bands and Dirac cones for freestanding silicene [5–7] seems to be confirmed also for silicene epitaxially grown on Ag(111) substrates. The stability and corresponding properties for germanium in the 2D pristine geometry, called germanene [5–7], have been theoretically studied. Tin also belongs to group IV of the periodic table and crystallizes at temperatures below 13.2 ° C in the diamond structure. Therefore, the existence of a 2D polymorph with honeycomb structure, called tinene, should be possible.
Among the unique properties of graphene, a constant infrared conductivity and optical absorbance limω→0A(ω) = πα with α = 1/137.076 as the Sommerfeld fine structure constant has been observed experimentally [1, 5] and explained in terms of non-interacting Dirac fermions with pseudospin [5, 8]. Recently, this result has been confirmed in the framework of independent Bloch–Fermi particles, optical interband transitions around the Dirac points, and a DFT description of the electronic structure [6, 7]. It could be demonstrated that the result A(0) = πα does not depend on the group IV atom, the sheet buckling, the hybridization state, and the details of the electronic structure around the Dirac points. Open questions, however, remain concerning the influence of the quasiparticle (QP) many-body effects due to optical excitation or relativistic effects in heavy atoms.
The quasiparticle effects significantly close the Dirac cones. In the graphene case, the corresponding Fermi velocity increases from the DFT value vF = 0.85 × 106 to 1.15 × 106 m s−1 including the excitation aspect [9]. A typical approximation for the description of the QP effects is the study of the exchange–correlation (XC) self-energy within Hedin's GW approximation [10] instead of the local or semilocal XC potential VXC in the Kohn–Sham equation of the conventional DFT. However, recently it has been proposed that QP effects on the electronic structure can be simulated to a large extent by means of spatially non-local potentials VXC obtained from hybrid XC functionals [11], such as the HSE06 one proposed by Heyd, Scuseria, and Ernzerhof [12, 13]. This approach is used in this work to study the influence of the excitation aspect on the electronic properties and optical absorbance.
Along the row C → Si → Ge → Sn the atomic weight increases from 12 to 119 suggesting a strong increase of relativistic effects on the motion of the electrons. Indeed, in the diamond structure with complete sp3 bonding for all group-IV materials, the spin–orbit splitting ΔSO of the p-like valence band states at the Γ point of the BZ increases. Values ΔSO = 6 (C), 44 (Si), 297 (Ge), and 770 (Sn) meV [14, 15] have been measured. While graphene without spin–orbit coupling (SOC) does not exhibit a bandgap at K or K', it does including SOC [16]. The corresponding energy dispersion deviates from those of Dirac cones and resembles that of massive relativistic particles. These effects are increased for silicene [17, 18] and are expected to be more pronounced for germanene and tinene. It has been argued that the resulting Dirac nature of the fermions together with the SOC-induced gap makes a topological insulator [18, 19]. In any case, the gap opening due to SOC should noticeably influence the absorbance in the infrared spectral region. Therefore, the impact of the SOC on A(ω) is studied in this paper. In contrast to the use of the XC self-energy [20] the SOC can be easily combined with the treatment of QP effects in the framework of hybrid functionals.
2. Methods
In our studies we employ density-functional theory (DFT) as implemented in the Vienna ab initio simulation package (VASP) [21] within the generalized-gradient approximation (GGA) of Perdew, Burke, and Ernzerhof (PBE) [22]. We account for quasiparticle effects describing exchange and correlation within the screened hybrid density functional HSE06 [12, 13, 23]. All-electron wavefunctions are generated within the projector-augmented wave (PAW) method [24], which allows for highly accurate optical transition matrix elements [25]. The energy cutoff Ecut for the plane-wave expansion of the wavefunctions between the cores depends on the atomic species in the system. After convergence tests we set Ecut = 400 eV (graphene), 245 eV (silicene), 175 eV (germanene), and 103 eV (tinene).
The 2D crystals are simulated within a supercell approach with L = 20 Å of vacuum in the direction perpendicular to the 2D crystal surface in order to avoid artificial interactions between the periodic images of the 2D sheet crystals. Spin–orbit interaction is taken into account. The ionic relaxation of all structures has been performed in the DFT-GGA framework until the Hellmann–Feynman forces are smaller than 0.001 eV Å−1. The hybrid functional HSE06 was used to calculate the electronic band structures and Pauli spinors at the obtained equilibrium geometries.
The optical absorption is studied in the framework of the independent-quasiparticle approximation [26] for the dielectric function. Whereas quasiparticle effects [10] are taken into account via the HSE functional, excitonic effects are not. These effects are more important in the energy range of the interband transitions [9]. Thereby, the complexity of the solution of the Bethe–Salpeter equation in the presence of SOC [20] can be avoided. Most convenient is the description of the optical transition matrix elements within the longitudinal gauge [25]. Then the influence of SOC on the optical oscillator strength and the inclusion of spin-flip transitions are easily described by the dipole matrix elements with Pauli spinors. For small frequencies, the calculation of the optical absorbance requires the use of a hybrid k-mesh strategy to sample the 2D BZ. Around the K and K' points a much finer k-point mesh has in fact been used. In total 128 × 128 Γ-centered k points in the 2D BZ of the crystals are used, whereas we applied a reduced grid of 32 × 32 q points for the calculation of the screened exchange interaction in the HSE functional [23]. Two additional refined grids are applied, each including 869 symmetry-reduced k-points within a circle of radius 0.05 × 2π/a and 0.005 × 2π/a, respectively, around one single K point in order to obtain the low-frequency limit of the absorbance. The method has been used successfully in conventional DFT calculations without SOC in previous works [6, 7].
3. Results and discussion
3.1. Geometries
It is well known that graphene is atomically flat, whereas silicene, germanene, and tinene should be buckled [27]. Using DFT-GGA for the ionic relaxation, we calculated the buckling height Δ and the in-plane nearest-neighbor distance d, which is related to the lattice constant a of the hexagonal lattice via 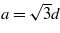 . The results are summarized in table 1. There are clear chemical trends. The characteristic lateral geometry parameters a and d and the buckling height Δ increase along the row C, Si, Ge, Sn. The increasing buckling amplitude indicates the deviation from the pure sp2 bonding and the formation of mixed sp2–sp3 bonding. The sp3 character also increases along the row C, Si, Ge, Sn. Indeed the buckling amplitude Δ approaches more and more the value
. The results are summarized in table 1. There are clear chemical trends. The characteristic lateral geometry parameters a and d and the buckling height Δ increase along the row C, Si, Ge, Sn. The increasing buckling amplitude indicates the deviation from the pure sp2 bonding and the formation of mixed sp2–sp3 bonding. The sp3 character also increases along the row C, Si, Ge, Sn. Indeed the buckling amplitude Δ approaches more and more the value 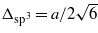 , 0.50 (C), 0.79 (Si), 0.82 (Ge), or 0.95 (Sn) Å, for complete sp3 bonding.
, 0.50 (C), 0.79 (Si), 0.82 (Ge), or 0.95 (Sn) Å, for complete sp3 bonding.
Table 1. Structural and electronic parameters. Electronic quantities are derived from hybrid HSE06 calculations. The GGA results are given in parentheses. Eg and m* are calculated with the inclusion of SOC (without SOC, Eg = m* = 0).
| C | Si | Ge | Sn | |
|---|---|---|---|---|
| a (Å) | 2.468 | 3.868 | 4.060 | 4.673 |
| d (Å) | 1.425 | 2.233 | 2.344 | 2.698 |
| Δ (Å) | 0.00 | 0.45 | 0.69 | 0.85 |
| vF (106 m s−1) | 1.01 (0.83) | 0.65 (0.53) | 0.62 (0.52) | 0.55 (0.48) |
| m* (m0) | 0.000 (0.000) | 0.001 (0.001) | 0.007 (0.008) | 0.029 (0.028) |
| Eg (meV) | 0.0 (0.0) | 1.9 (1.5) | 33 (24) | 101 (73) |
3.2. Quasiparticle band structures
The band structures obtained from Kohn–Sham eigenvalues of the DFT are well known for graphene, silicene, and germanene [7, 9, 27]. For tinene the bands  ν(k) are plotted in figure 1. Within the DFT-GGA framework, tinene shows a (zero-gap) semiconducting character in contrast to the α-Sn crystallizing in diamond structure [28] with a L6c band below the Fermi level. The apex of the valence band Dirac cone around K and K' points in tinene fixes the Fermi energy as a shortcoming of the method. Also with the HSE06 functional, we find a zero-gap semiconductor, but with an increased direct bandgap at the Γ point. For all 2D crystals under investigation, the hybrid functional HSE06 without spin–orbit coupling (SOC) leads to zero-gap semiconductors with isotropic Dirac cones at the K and K' points of the hexagonal BZ. Only the band dispersion is modified with respect to the GGA calculation. In table 1 we compare the Fermi velocities obtained with both functionals for the four 2D honeycomb crystals. The Fermi velocity decreases along the row C, Si, Ge, and Sn independent of the functional. The quasiparticle corrections are zero at the Fermi level [10] thus, in zero-gap semiconductors, quasiparticle corrections do not open a bandgap. Only the dispersion of the conduction and valence bands increases and, consequently, leads to an increase of the Fermi velocity when using HSE06 compared to DFT-GGA (see figure 1 for tinene). The Fermi velocity of freestanding graphene has been determined experimentally. Its value is about 1.1 × 106 m s−1 [29], and thus well predicted by the HSE06 functional (see table 1). The QP value vF = 1.15 × 106 m s−1 using the GW approximation is slightly larger [9]. Larger values measured by means of angle-resolved photoemission spectroscopy (ARPES) for silicene [2] than those in table 1 are probably a consequence of the substrate influence. We conclude that the HSE functional simulates important features of the QP approximation. The energy gaps outside K or K' are significantly opened while the wavevector dispersion near K or K' is increased with respect to standard semilocal DFT calculations.
ν(k) are plotted in figure 1. Within the DFT-GGA framework, tinene shows a (zero-gap) semiconducting character in contrast to the α-Sn crystallizing in diamond structure [28] with a L6c band below the Fermi level. The apex of the valence band Dirac cone around K and K' points in tinene fixes the Fermi energy as a shortcoming of the method. Also with the HSE06 functional, we find a zero-gap semiconductor, but with an increased direct bandgap at the Γ point. For all 2D crystals under investigation, the hybrid functional HSE06 without spin–orbit coupling (SOC) leads to zero-gap semiconductors with isotropic Dirac cones at the K and K' points of the hexagonal BZ. Only the band dispersion is modified with respect to the GGA calculation. In table 1 we compare the Fermi velocities obtained with both functionals for the four 2D honeycomb crystals. The Fermi velocity decreases along the row C, Si, Ge, and Sn independent of the functional. The quasiparticle corrections are zero at the Fermi level [10] thus, in zero-gap semiconductors, quasiparticle corrections do not open a bandgap. Only the dispersion of the conduction and valence bands increases and, consequently, leads to an increase of the Fermi velocity when using HSE06 compared to DFT-GGA (see figure 1 for tinene). The Fermi velocity of freestanding graphene has been determined experimentally. Its value is about 1.1 × 106 m s−1 [29], and thus well predicted by the HSE06 functional (see table 1). The QP value vF = 1.15 × 106 m s−1 using the GW approximation is slightly larger [9]. Larger values measured by means of angle-resolved photoemission spectroscopy (ARPES) for silicene [2] than those in table 1 are probably a consequence of the substrate influence. We conclude that the HSE functional simulates important features of the QP approximation. The energy gaps outside K or K' are significantly opened while the wavevector dispersion near K or K' is increased with respect to standard semilocal DFT calculations.

Figure 1. Band structure of tinene with SOC computed with a semilocal GGA XC potential (blue dashed lines) and a non-local XC potential derived from the HSE06 hybrid functional (red solid lines). The energy zero is defined by the Dirac points. The Fermi level is indicated by a horizontal line.
Download figure:
Standard image High-resolution imageWe investigated the influence of SOC on the quasiparticle band structure, in particular close to the Dirac points. This requires much numerical effort. The convergence of the electronic bandgap induced by SOC as a function of the number of k points is very slow when using hybrid functionals. The regular 2D k-point meshes have to be carefully chosen since the convergence is strongly enhanced if the K and K' points are excluded from the regular k-point mesh [30]. Consequently, we applied a 32 × 32 × 1 Γ-centered Monkhorst–Pack k points mesh [31] for all materials. The HSE06 band structures of the four 2D honeycomb crystals along high-symmetry lines in the hexagonal BZ are plotted in figure 2. For germanene and tinene the inset also shows the linear dispersion around the K or K' point obtained without SOC in comparison to the dispersion with SOC. It can be seen that SOC induces a fundamental gap Eg and, hence, massive Dirac particles with effective masses m* close to the conduction and valence band edges near K (or K') points. We do not find a k-induced splitting of the twofold degeneracy due to SOC, i.e. away from Γ. At least along the high-symmetry lines we do not observe Rashba [32] or Dresselhaus [33, 34] components in the band structure, in agreement with other studies [17, 18]. At the Γ point a similar splitting of the fourfold degenerate uppermost valence bands into two twofold degenerate valence bands occurs as in the bulk. For graphene, the smallness of the SOC-induced Eg makes its precise HSE06 computation difficult. Since the size of the estimated bandgap Eg is about 0.02 meV, we only discuss the effect of SOC for the other group IV materials. The electrons ν =+ and holes ν =− in silicene, germanene, and tinene in the vicinity of K or K' can be approximately described by a dispersion relation of massive relativistic Dirac particles [35]
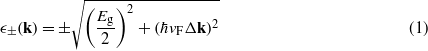
with Δk as the deviation of the 2D wavevector from its value at a K or K' point. The SOC-induced gaps Eg are also listed in table 1. They follow the same clear chemical trend as the spin–orbit splittings ΔSO in the diamond structures of the group IV materials mentioned above. The Eg values are, however, by one order of magnitude smaller. Thereby, the spin–orbit effects using non-local HSE06 XC potentials are significantly larger than those obtained within DFT-GGA as a consequence of the more localized quasiparticle states. Our findings in table 1 are in agreement with Eg values in other recent DFT computations. Values Eg = 1.55 (Si), 23.9 (Ge), and 73.5 (Sn) meV have been given in the literature [16, 35, 36]. The finite gap gives rise to massive Dirac (electron and hole) particles near a K or K' point with the effective mass

Its values in table 1 also show a clear increase with the atomic number of the group IV element.

Figure 2. Quasiparticle band structures of (a) graphene, (b) silicene, (c) germanene, and (d) tinene along high-symmetry lines of the 2D hexagonal BZ using the HSE06 functional including spin–orbit interaction. For germanene and tinene, the inset shows a magnification of the electronic bandgap around the Dirac point together with the band structure obtained without SOC (solid black lines).
Download figure:
Standard image High-resolution image3.3. Infrared absorbance
The absorbance of group IV honeycomb crystals exhibits the peculiarity that the optical absorption is related to the fine structure constant independent of the group IV atom, the sheet buckling, and the orbital hybridization in the limit of vanishing photon frequencies [6, 7]. On the other hand, one knows that semilocal DFT functionals rather accurately predict ground state properties, but fail to predict excited state properties such as the optical bandgap in non-metals and the correct frequency dependence of the dielectric function. However, we deal here with zero-gap semiconductors (at least without SOC). Since in zero-gap semiconductors with Dirac cones only the Fermi velocity changes due to quasiparticle effects, it should be possible to determine the optical absorbance in the low-frequency regime without applying more sophisticated approaches to the electron–electron interaction, such as the many-body perturbation theory within the GW approximation, or the Bethe–Salpeter equation to account for excitonic effects.
In contrast, the increasing importance of SOC on the electronic properties for heavy group IV atoms and the deformation of the Dirac cones near the K and K' points ask for a more detailed investigation of the impact of the SOC on the optical properties, therefore, we studied the optical absorbance A(ω). This is connected to the frequency-dependent imaginary part of the dielectric function ε(ω) of the 3D supercell with lattice constant L by [7]

The optical absorbance resulting from the independent-quasiparticle computations for all four group IV honeycomb crystals as a function of the photon energy is shown in figure 3. Without SOC the low-frequency absorbance approaches the well-known limit A(ω → 0) = πα as demonstrated earlier for independent Kohn–Sham electrons [7]. However, using the HSE approach, the optical absorption peaks are blueshifted due to quasiparticle corrections. In a wide frequency range the optical absorbance taking into account SOC shows a very good overall agreement with the absorbance obtained without SOC. However, a more detailed analysis reveals a remarkable modification of the optical absorbance close to the fundamental absorption edge. For ħω < Eg the absorption vanishes, while for ħω > Eg the absorbance is significantly increased for ħω → Eg. This behavior is particularly pronounced for germanene (figure 3(c)) and tinene (figure 3(e)). Within our numerical accuracy we estimate that the absorbance is increased by a factor of two at ħω = Eg due to SOC.

Figure 3. Optical absorbance in units of πα as a function of the photon energy for (a) graphene, (b) silicene, (c) germanene, and (d) tinene without (black solid lines) and with (red solid lines) spin–orbit coupling. The inset in (c) and (d) depicts the region around their respective fundamental absorption edges.
Download figure:
Standard image High-resolution imageThis peculiar lineshape of the absorbance in the vicinity of the fundamental absorption edge asks for a detailed explanation. In general, it is a consequence of the relativistic band dispersions (1) and the optical matrix elements (see equation (7) in [7]) modified due to the inclusion of SOC when calculating the imaginary part of the dielectric function. The modified band dispersion is illustrated in figure 2. The strength of the corresponding interband transitions is displayed in figure 4. As an example, we study the optical matrix elements between the highest occupied and the lowest unoccupied Bloch bands of germanene along the two high-symmetry directions KΓ and KM. The optical transition matrix elements resulting within the longitudinal approximation for Pauli spinors and germanene near a K or K' point are displayed in figure 4 for in-plane light polarization.

{kind=link}
{kind=link}
{kind=link}
Figure 4. A normalized optical matrix element of germanene without (black solid line) and with (red solid line) spin–orbit interaction between the highest occupied and lowest unoccupied Bloch bands in the vicinity of K. At K, without SOC, the normalized optical matrix element approaches the ideal value 1.
Download figure:
Standard image High-resolution image{kind=link}
Without SOC, the squares of the momentum-matrix elements D(k) = ∑j=x,y|〈ck|pj|vk〉|2 reduce to 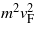 at a Dirac point
at a Dirac point  with a weak, almost linear, Δk dependence, whereas the matrix elements D(k) including SOC are significantly enhanced by a factor of two near
with a weak, almost linear, Δk dependence, whereas the matrix elements D(k) including SOC are significantly enhanced by a factor of two near  with a pronounced k dependence. Further away from the Dirac point the matrix elements with and without SOC are practically identical.
with a pronounced k dependence. Further away from the Dirac point the matrix elements with and without SOC are practically identical.
With SOC the matrix elements display an additional Lorentzian Δk dependence near a Dirac point. This can be represented analytically by
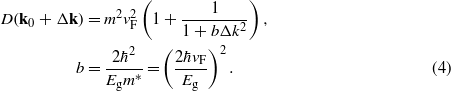
The quantity b is determined by exhausting the oscillator-strength sum rule on the dielectric function in the low-frequency regime. We have to stress that the agreement of equation (4) with the numerical curve in figure 4 is excellent. The same holds for silicene and tinene (not shown). Including the corrected matrix elements (4) the optical absorbance is readily calculated by means of the analytical method described in a previous work [7]. Taking into account the modified band dispersion equation (1) due to the massive Dirac particles in the presence of SOC we derive
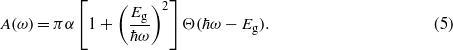
Since the joint density of states near the Dirac points ∼ħωΘ(ħω − Eg) is still linear in the photon energy, the strong modification of the absorbance is mainly a consequence of the modified transition matrix elements (4). The result recovers important features encountered in the numerical calculations. First of all, for photon energies that are large (but much below the optical interband transitions near critical points in the band structure in figures 1 and 2) compared to the electronic bandgap, the absorbance reduces to the well-known limit A(ω ≫ Eg/ħ) = πα. Furthermore, for photon energies in the vicinity of the fundamental absorption edge the absorbance increases to A(ω ≈ Eg/ħ) = 2πα. In between these two limits the absorbance follows the power law as predicted by equation (5). For vanishing SOC, i.e. Eg → 0, the previous analytical description is recovered.
The previous findings, in particular equations (4) and (5), also hold for GGA calculations, if the proper band gaps Eg and Fermi velocities vF are taken from table 1.
4. Conclusion
We have studied in detail the impact of quasiparticle and spin–orbit effects on the electronic and optical properties of the group IV honeycomb crystals graphene, silicene, germanene, and tinene. Many-body effects are incorporated in the framework of the non-local exchange–correlation functional HSE06. While both many-body and relativistic effects modify the electronic structure near the Dirac points, the modification of the optical absorption is restricted to small frequencies near the SOC-induced fundamental gap. This gap leads to Dirac particles with a finite mass. The excitation effects mainly modify the Fermi velocity vF. Their influence on the particle mass and the gap Eg is, however, small.
Most important for the low-frequency absorbance is the SOC-induced gap Eg. The bandgap increases along the row C → Si → Ge → Sn and is larger in HSE than that obtained with standard DFT functionals. The band dispersion represented by vF is of minor influence. The finiteness of Eg gives rise to a drastic modification of the joint density of states as well as the optical transition matrix elements for small photon energies and, hence, close to a Dirac point. No absorption happens for ħω < Eg, while the absorbance A(ω = Eg/ħ + 0+) = 2πα near the gap is increased. In the frequency region between gap and optical interband energies the absorbance value A(ω) = πα without SOC is recovered. The results are independent of the group IV honeycomb structure, but more pronounced for heavier elements.
Acknowledgments
LM acknowledges financial support from the Carl-Zeiss Foundation and the DFG Graduate School 1523 'Quantum and Gravitational Fields'.