
Abstract
MgTFSI2 is the only ether-soluble "simple" magnesium salt. The poor electrochemical performance of Mg electrodes in its solutions hinders its practicality as a viable electrolyte for Mg batteries. MgTFSI2/DME solutions were demonstrated to dissolve large quantities of MgCl2 and produce electrolyte solutions with superior performance, though the electrochemical performance, mainly in terms of reversibility, of MgTFSI2/MgCl2 (DME) solutions cannot yet compete with that of organometallic based electrolyte solutions. We believe that the solutions' purity level governs the overall electrochemical performance, especially in solutions where a strong reductant (i.e Grignard reagent) is not present to act as an impurity scavenger. In this work, we alter the performance of the MgTFSI2/MgCl2 (DME) solutions through chemical and electrochemical conditioning and demonstrate the effect on the solutions' electrochemical characteristics. We demonstrate relatively high reversible behavior of Mg deposition/dissolution with crystalline uniformity of the Mg deposits, complemented by a fully reversible intercalation/de-intercalation process of Mg ions into Mo6S8 cathodes. We also investigated LiTFSI/MgCl2 solutions which exhibited even higher reversibility than MgTFSI2/MgCl2 (DME) solutions, which we attribute to the higher purity level available for the LiTFSI salt.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 License (CC BY, http://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse of the work in any medium, provided the original work is properly cited.
Magnesium is a natural candidate anode material for "next generation" rechargeable batteries due to its high volumetric capacity (3833 mAh/cm3), low reduction potential (−2.3 V) wide abundance, and low price.1 Rechargeable Mg battery research had been developing very slowly since the 1920's but had recently gained a big momentum. Magnesium battery systems will have great difficulties to outperform lithium systems In terms of energy and power density. However, they possess several properties that make them desirable, as they are expected to benefit from a cheaper price and lower hazard levels. One of the core issues developing rechargeable magnesium batteries is the formulation of electrolytic solutions that support reversible magnesium deposition. Other properties such as sufficient ionic conductivity, adequate magnesium ion concentration, and a wide electrochemical window are also mandatory. 15 years ago we synthesized electrolyte solutions that possessed most of the key features listed above. These electrolyte solutions were the product of a Lewis acid/base reaction in which R2Mg moieties such as Bu2Mg served as the base component, and RAlCl2 species such as EtAlCl2 served as the acid component. Hence, we could demonstrate a family of organometallic electrolyte solutions for rechargeable Mg batteries known as di-chloro complex solutions (DCC).2,3 Unfortunately, even the best DCC electrolyte solution does not possess the minimum requirement needed for next generation rechargeable magnesium batteries, e.g. wide electrochemical stability window (>2.2 V), chemical stability and safety. The use of aromatic ligands enabled to develop electrolyte solutions with wider electrochemical windows > 3 V.4 It should be noted that in recent years, several families of electrolyte solutions for Mg batteries which exhibit wide electrochemical windows were developed by other groups.5–11 Recently we demonstrated organometallic-free electrolyte solutions named MACC. These solutions were comprised of the reaction products between MgCl2 and AlCl3 in THF.12 Although these solutions support reversible magnesium deposition, their high concentration of reactive chloride-containing species exhibit detrimental properties which render them unfeasible for good batteries application. Their main problem is incompatibility with any transition metal oxides that can be used as high voltage cathodes for the next generation of Mg batteries (e.g. V2O513,14). In order to develop electrolyte solutions with all of the desired properties listed above, new concepts are required with regard to both the solvents and the salts. The salts to be used should contain no organometallic compounds, namely, their anions should be similar to those that are ubiquitous in the Li battery world e.g.: Mg(BF4)2, Mg(ClO4)2 and Mg(TFSI)2. Unfortunately, most of such salts are insoluble in ethereal solvents, which are currently the only viable solvents to be used, due to their inertness with Mg metal. Mg(TFSI)2 (magnesium bis trifluoromethanesulfonyl imide) is the only readily ether-soluble salt (which answers the definition above), possessing good ionic conductivity, wide electrochemical stability window and an exceptional chemical stability. Therefore it has been getting great attention lately.15–17
Preliminary experiments with ethereal solutions of pure MgTFSI2 have shown that it does not support stable and reversible magnesium deposition. In turn, recent studies have demonstrated that addition of chlorides to the MgTFSI2 pure solutions (in DME) yields solutions that support highly reversible and stable magnesium deposition.18 Availability of such solutions in which Mg electrodes behave reversibly, may be revolutionary for developing the next generation of rechargeable Mg batteries. Thereby, these solutions deserve serious and systematic studies, in order to properly understand their behavior. This paper reports on such studies.
Experimental
Solution preparations
LiTFSI/MgTFSI2 solutions were prepared as follows: predetermined amounts of LiTFSI/MgTFSI2 and MgCl2 were added to DME/Diglyme/Triglyme (Dimethoxyethane) and stirred for 6 h at 70 °C. The solutions were then filtered. LiTFSI (99.95%), MgCl2 (99.99%), DME, Diglyme, Triglyme, and DiButhyl-magnesium were obtained from Sigma-Aldrich. MgTFSI2 (97%) and MgTFSI2 (99.95%) were purchased from Strem/Alfa-Aesar and Solvonic respectively (MgTFSI2 exhibited similar performance for all vendors). MgTFSI2 and LiTFSI were dried under vacuum for 24 h at 250 °C and 120 °C respectively prior to synthesis.
Measurement conditions
All electrochemical measurements and solution preparations were performed in a 99.999% Ar filled glove box.
Cell configuration and materials
All electrochemical measurements were performed in flooded 3 electrode glass cells. Mg foil/strips were used as the counter/reference electrodes in all measurements. Pt was used as the working electrode for all metal deposition/dissolution measurements. V2O5 thin film electrodes were prepared by thermal vacuum deposition from a V2O5 powder (Sigma Aldrich). Film thickness was usually around 200 nm. Electrodes were then annealed at 415 °C for 4.5 h under air to obtain the desired crystalline phase.
Electrochemical measurements were performed with a PAR 273A potentiostat, except for the Mo6S8 intercalation measurement (Fig. 10a) which was performed with a VMP-2 potentiostat, Bio-Logic Co. Electrochemical conditioning of MgTFSI2 based solutions was comprised of a cyclic voltammetry performed on a Pt working electrode between −0.8 V and 2.0 V at 1 mV/sec. For macro-reversibility measurements ca 1 micrometer Mg (0.5 coulombs/cm2) was deposited at 1 mA/cm2. Then, 20% of the initially deposited film was electrochemically dissolved and re-deposited at current density of 1 mA/cm2. After 100 cycles (in relevant cases), the remaining Mg deposit was electrochemically dissolved at 1 mA/cm2 until the voltage steeply rose, indicating the end point. Cycling efficiency is calculated from the final and initial charges for Mg deposition and dissolution, respectively, divided over the number of full cycles. Cycling efficiency for macro-reversibility measurements were calculated with the following formula:
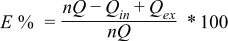
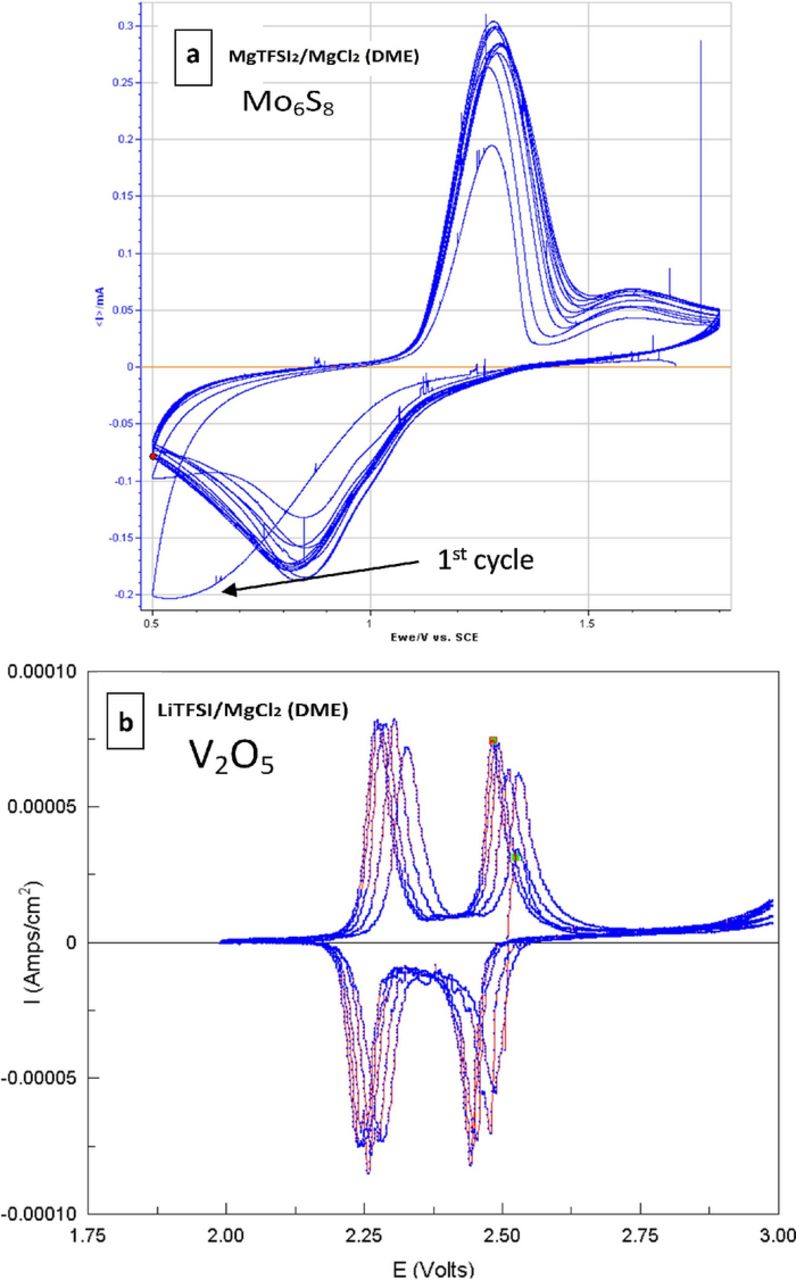
Figure 10. (a) Repeated cyclic voltammograms of composite Chevrel phase electrodes (85% Mo6S8, 10% carbon black, 5% PVDF) which undergo repeated intercalation/de-intercalation processes with Mg2+ ions in MgTFSI2 0.25 M + MgCl2 0.5 M/DME solution, 0.1 mV/sec. (b) Repeated cyclic voltammograms for Li+ intercalation into a V2O5 thin film electrodes in LiTFSI 0.5 M + MgCl2 0.125 M/DME solution, 0.1 mV/sec.
Where Q is the charge cycled, Qin is charge of the Mg at the beginning of the experiment and the Qout is the residual Mg charge at the end of the experiment.
Electrochemical quartz crystal microbalance (EQCM) measurements were performed with an SRS QCM200 quartz crystal microbalance controller using a 5 Mhz AT-cut Pt plated quartz crystal.
Morphology
Images and elemental analysis were carried out with Helios 600 scanning electron microscope system (FEI). Substrates for morphological study were comprised of 100 nm sputtered Pt glass slides.
XPS Analysis
XPS analyses were performed in a Kratos AXIS-HS spectrometer, using a monochromatized Al Ka source. Survey and high resolution scans were acquired at 150 W. All acquisitions were performed in a hybrid mode (using electrostatic and magnetic lenses) and detection pass energies of 80 or 40 eV for survey and high resolution scans, respectively. All acquisitions were made with low energy electron flood gun for charge neutralization. Samples were transferred from the glove box to the XPS load-lock under inert Ar atmosphere using proprietarily made, air-tight, transfer system.
All XPS measurements were carried out at room temperature, under vacuum conditions of (1.0–3.0) 10–9 Torr. The spectrometer energy scale was routinely calibrated according to the ISO TC/201 SC7 international procedure for binding energy (BE) with Au 4f7/2) 83.98 and Cu2p3/2 ) 932.67. Data processing was done with either VISION2.1 software ("Kratos"). In most cases a Shirley background was used. Curve fitting were performed using a 80/20 Gaussian/Lorentzian line shape .Regularly, 100–1000 iterations were used to reach best fit.
Results and Discussion
The electrochemical behavior of crude MgTFSI2 solutions in DME, without and with MgCl2
Electrolyte solutions consisting of pure MgTFSI2 salt in DME show a rather poor performance in connection with Mg electrochemistry. A typical Cyclic voltammetry measured with an inert Pt WE in MgTFSI2 0.5 M (in DME) solutions (Fig. 1a), exhibits an ever increasing negative current, when the electrode's voltage is scanned negative to the Mg RE [reference electrode], at relatively higher over-potentials (−0.6 V Vs Mg) than what is expected for normal magnesium deposition. On the scan back, positive currents are detected only at a very high over-potential of 1.5 V vs. Mg. The reversibility in terms of charge for the oxidation/reduction processes is around 60%, which is quite low. We attribute the cathodic process to magnesium deposition and these observations may mean that quasi-passivating layers are formed on any freshly deposited magnesium metal in these solutions.
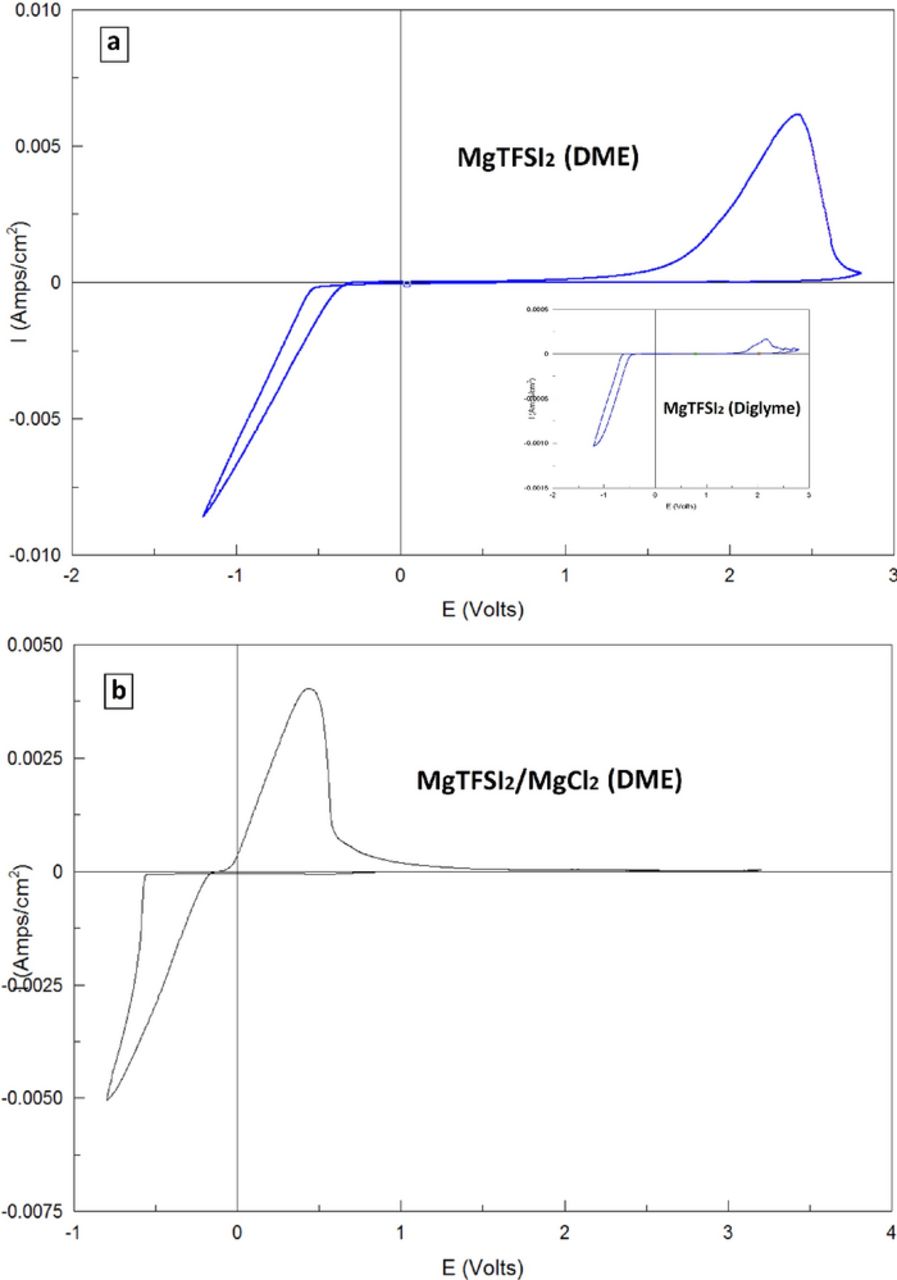
Figure 1. A typical cyclic voltammetry (CV) of Pt working electrodes in (a) MgTFSI2 0.5 M in DME solution, Scanned at 25 mV/sec. The inset shows a CV of Pt electrode in MgTFSI2 0.5 M solution in diglyme. (b) MgTFSI2 0.25 M with MgCl2 0.5 M in DME. Mg foils served as reference and counter electrodes RE, CE), scanned at 25 mV/sec. Addition of MgCl2 significantly reduces the overpotential for both deposition and dissolution processes.
A Recent report has shown that reacting MgTFSI2 with MgCl2 in DME yields solutions which support highly reversible Mg deposition.18 We specifically use the term "reacting" since simple dissolution and mixing cannot explain high solubility of the nearly insoluble MgCl2 (in pure DME) when it comes to MgTFSI2/ DME solutions.
Interestingly, the electrochemical response measured for the MgCl2 containing solutions (Fig. 1b) is strikingly different than that for the MgTFSI2/DME solution. Similarly to the MgCl2-free solutions, in MgCl2-containing MgTFSI2/DME solutions, appreciable negative currents are seen on the negative scan at overpotential of 0.6 V vs. Mg RE. Most noticeable however, is that in these solutions, the positive scan is characterized by a large positive peak, beginning just after crossing 0 V vs. Mg, the point expected for commencing magnesium dissolution. Its magnitude was found to be directly related to the amount of charge that passed on the negative scan.
Since MgCl2 is insoluble in ethereal solutions. We hypothesize at this early point that the two salts react in a Lewis acid-base reaction to form various electrochemically active Mg/Cl/TFSI complexes, through a ligands exchange reaction. We also hypothesize that the evolved chloride ligands containing species play a crucial role in defining the properties of this system. We believe that the corrosive nature of these anions, along with the slight solubility of MgCl2 in many organic solvents, creates the needed conditions for some surface films instability. Previous studies with organometallic type solutions comprising ether solvents, MgR2 type Lewis bases and AlCl3-nRn type Lewis acids have shown that the higher the total amount of chlorides in the system, the higher is the overpotential for Mg deposition and dissolution. One explanation for this phenomenon assumes that chloride anions and chloride containing species adsorb strongly to the electrode surface compared with organic ligands. Such adsorption may inhibit possible surface reactions of magnesium electrodes with any reactive solution species and avoid their passivation. However, the presence of Cl− anions is expected to come with a penalty of surface resistance, higher over-potential for Mg deposition and detrimental interactions with transition metal oxides that can serve as Mg ions insertion cathodes.
Higher glymes such as diglyme and triglyme were screened to replace DME as the solvent. MgTFSI2 freely dissolves in both diglyme and triglyme, and exhibits similar electrochemical performance to DME based solutions (Fig. 1a inset). MgCl2 has a very poor solubility in MgTFSI2/Diglyme solutions and only moderate solubility in MgTFSI2/triglyme solutions (only up to 0.25 M of MgCl2 can be dissolved in a 0.25 M MgTFSI2/triglyme solution, while over 0.5 M of MgCl2 can be easily dissolved in a 0.25 M MgTFSI2/DME solution). A solution consisted of MgTFSI2 (0.25 M) + MgCl2 (0.25 M) in triglyme exhibited no peaks that are associated with Mg deposition/dissolution processes. Hence, DME so far is the most adequate solvent to be used with MgTFSI2/MgCl2 solutions in connection to reversible Mg deposition.
MgTFSI2/LiTFSI/MgCl2 solutions conditioning and electrochemical analysis
Although, the addition of MgCl2 to MgTFSI2/DME solutions leads to possible reversible Mg deposition, the level of reversibility is rather poor: cannot exceed 80%-90%, with further deterioration observed during consecutive cycles. We should mention that MgTFSI2 based solutions are susceptible to impurities. In turn, organo-metallic based (or even Boro-Hydride based9,11) Mg salts solutions in ethers are reductive in nature and able to eliminate most of the reactive impurities in non-aqueous solutions, including trace water, oxygen, CO2 and protic acids. Thereby, it is easy to obtain bare, surface films (and passivation) free Mg electrodes in the organometallic type complex solutions and hence obtain fully reversible Mg deposition/dissolution processes.
Previously, we have demonstrated a procedure, we termed "conditioning" to enhance the electrochemical performance of MACC solutions.19 Although we did not have enough evidence to determine why the process has such an enormous effect on the electrochemical performance, we strongly believe that the elimination of impeding impurities during the process is a major reason. Recently, Gewirth et al. published a paper proposing an additional mechanism for such "conditioning" processes which has a chemical nature.20 Interestingly, we have discovered that the conditioning process, previously used on MACC solutions, has the same effect on the electrochemical performance of MgTFSI2/MgCl2 (DME) solutions. Figure 2a shows consecutive voltammograms of a Pt electrode in a MgTFSI2 (0.25 M) + MgCl2 (0.5 M) (DME) solution during the conditioning process. The conditioning process consists of cycling at 1 mV/sec between −0.8 V and 2.0 V. During consecutive cycles, an enhancement in the reversibility of the Mg deposition process is clearly observed, expressed by lower over-potentials and higher cycling efficiencies. These potential limits (−0.8 V, 2.0 V) were found to result in consistently well behaving, conditioned solutions. Although we do not fully understand the conditioning process, we do know that the conditioning process requires the passage of reductive faradaic currents associated with substantial charge passage, and therefore the negative scan should be span beyond the overpotential for magnesium deposition.
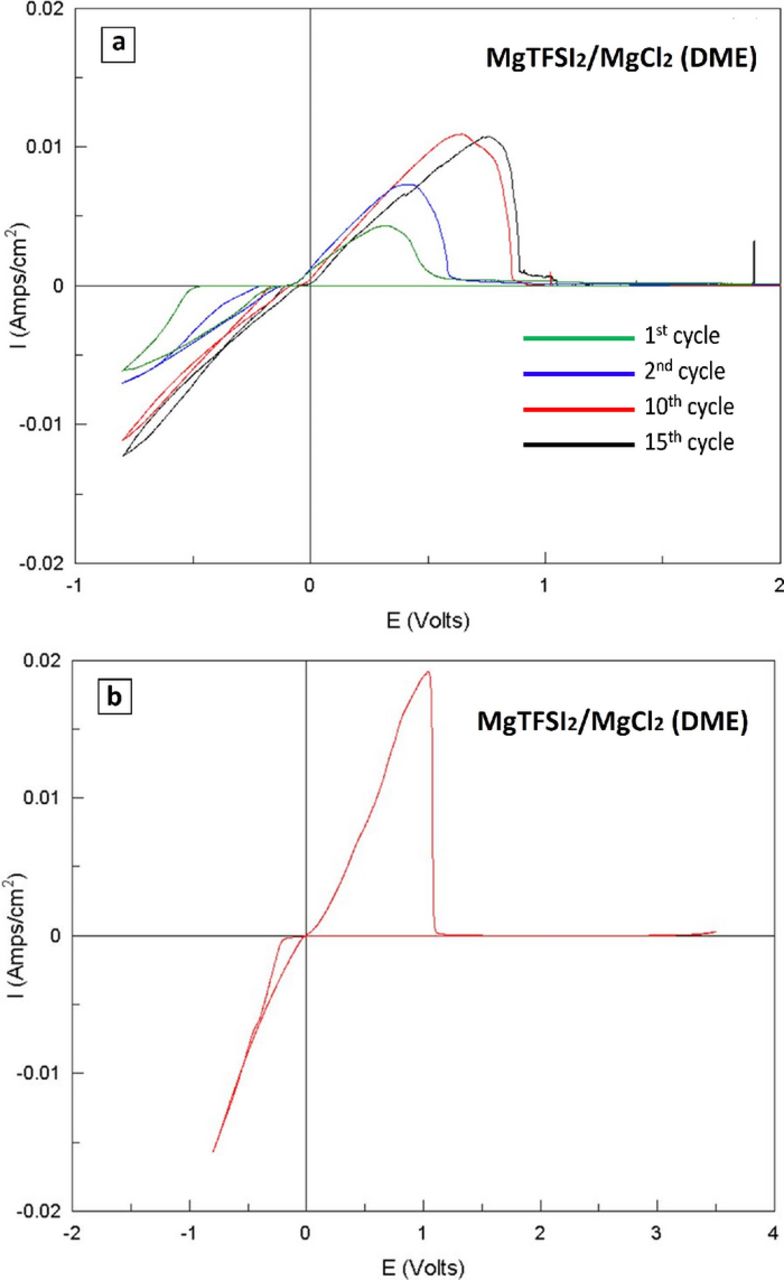
Figure 2. (a) Selected CV cycles during the conditioning procedure of MgTFSI2 0.25 M with MgCl2 0.5 M in DME solution. Pt served as the WE, and Mg as the RE and CE, scan rate 1 mV/sec. (b) A typical cyclic voltammogram of Pt WE in a conditioned MgTFSI2 0.25 M + MgCl2 0.5 M in DME solution, Mg foils served as the RE and CE, scan rate 25 mV/sec.
Following the conditioning process, the solution retains its newly obtained enhanced electrochemical properties when introduced to a fresh cell with fresh electrodes. A typical voltammogram of a solution that has undergone conditioning is shown in Figure 2b (obtained in a fresh cell with fresh electrodes). The post-condition solution exhibits 98% reversibility, over-potential for deposition of 0.2 V and an electrochemical window of 3.1 V, which reflect the normal anodic stability of ether solvents. Fig. 3 shows the frequency/mass response in an EQCM measurement during cyclic voltammetry. The mass change during the Mg deposition/dissolution processes correlates with the charge transferred during the measurement: all the mass deposited during the cathodic process is fully dissolved during the corresponding anodic process, fully in line with Mg deposition process with cycling efficiency approaching 100%. However, in all of these EQCM experiments, the mass per electron (m.p.e.) value calculated during the measurements was lower than 12 (the equivalent weight of magnesium). For instance, the m.p.e. calculated for the experiment related to Figure 3 was 9.39. Based on parallel studies of complex electrochemical processes by EQCM,21–23 we can attribute such a discrepancy to viscoelastic effects that limit the simple applicability of the Sauerbrey equation24 for these measurements.
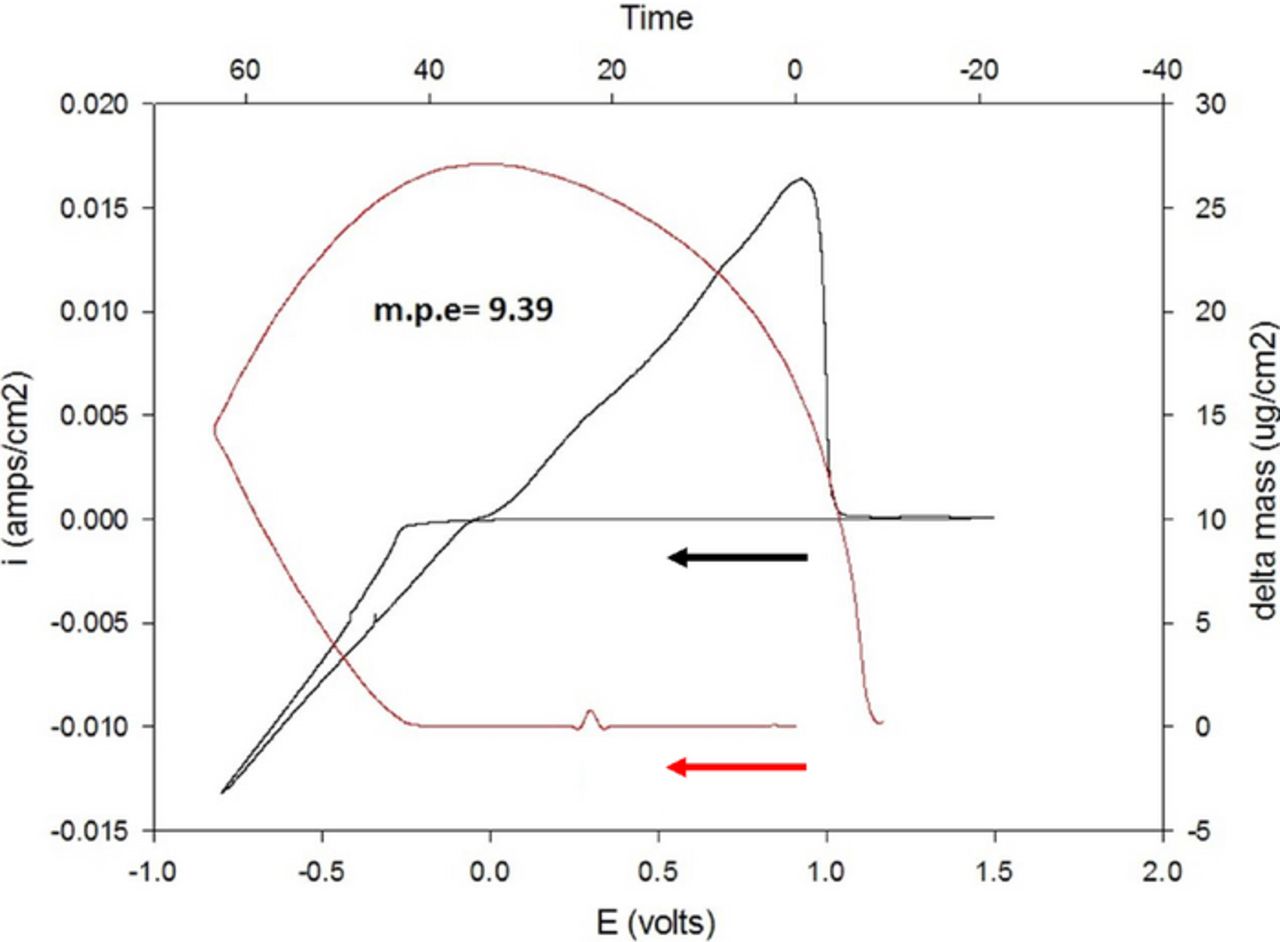
Figure 3. Typical results of EQCM experiments (simultaneous CV and mass accumulation/depletion response) with Pt electrode plated on 5 MHz AT cut quartz crystal in a MgTFSI2 0.25 M + MgCl2 0.5 M in DME solution. The black line represents a cyclic voltammetry measured at a scan rate of 25 mV/sec. The red line represents the change in mass during cycling (translated from frequency changes, based on the Sauerbrey equation24). Mg foils served as the RE and CE.
All our tests with MgTFSI2/MgCl2 solutions showed that during prolonged electrochemical measurements, an un-identified by-product was formed (and at least part of it precipitated on the electrode surface), a by-product which is associated with the passage of faradaic currents. The by-product has a distinctive black color and it readily dissolves in the solution, resulting in black murkish colored solution. We attributed this at first to the purity of our procured MgTFSI2 source (all vendors we tried led to the same result. Alfa Aesar, Strem, Solvonic). We have therefore substituted MgTFSI2 with LiTFSI, as LiTFSI is normally obtained at much higher purity levels (and much cheaper price). Our working hypothesis is that the reaction between MgTFSI2 and MgCl2 leads to the formation of the active species, e.g. DME complexed MgCl+ and/or Mg2Cl3+. Hence, substituting for LiTFSI should yield a very similar "model" system. MgCl2 dissolves In LiTFSI (DME) solutions although a higher concentration of LiTFSI is required to form clear solutions. Prolonged electrochemical measurements with LiTFSI/MgCl2 (DME) in most cases did not seem to produce any visible by-products and the solution remained clear and colorless.
One of the advantages of using LiTFSI with MgCl2 as raw materials is associated with the availability of highly pure and super dry raw materials. Very pure and dry MgTFSI2 is hard to procure, and is very expensive. On the contrary, LiTFSI and MgCl2, at the highest purity and dryness are offered by many manufacturers at reasonable price. Due to the extreme sensitivity of metallic magnesium to water, very dry solutions are mandatory for good performance.
Preliminary tests have shown that optimal solutions are produced by reacting 0.5 M LiTFSI with 0.125 M MgCl2 (in DME). This formulation yielded clear solutions with preferred electrochemical performance (after a "conditioning process"). Formulations that yielded turbid solutions or too high Li concentration were not used. Initial CV measurements with the above solution gave poor results, as can be seen from Fig. 4 (red curve). No clear reduction/oxidation peaks related to Mg deposition/dissolution processes were observed. We have once again implemented electrochemical manipulation to enhance the electrochemical performance. However in the case of LiTFSI based solutions we have found that the same potential sweep that enhanced the electrochemical performance of MACC and MgTFSI2 based solutions, seemed to have a much smaller effect on LiTFSI/MgCl2 solutions, resulting in a very long pretreatment process. Galvanostatic cycling (see methods), in the case of LiTFSI solutions, produced much better results. Nonetheless, we believe that both processes (galvanostatic cycling and CV) are similar in nature and affect the respective solution in a similar manner; we will regard both processes as "conditioning". The galvanostatic conditioning process included a repeated passage of constant charge at constant current, following the over-potential of the cathodic process (Mg deposition). Initially, in a fresh LiTFSI/MgCl2 solution, the electrochemical response exhibited a very high over-potential for deposition, of ca. 0.8 V for the first few cycles. Upon cycling, the over-potential for Mg deposition decreases monotonically from cycle to cycle, until it stabilized at a low value < 0.2 V that indicates that the solution reached an optimal composition (i.e. became pure enough to allow fully reversible Mg deposition). The optimal behavior of the DME/LiTFSI(0.5 M)/MgCl2(0.25 M) solutions is reflected by the voltammetric behavior seen in Fig. 4 (blue curve). The CV presented (obtained with a pretreated solution in a fresh cell), exhibits low Mg deposition overpotential, of less than 0.2 V, Mg deposition/dissolution efficiency >99% and a wide electrochemical window >3.1 V. These findings prove that this "conditioning" procedure affects only the solution, and it is not associated with any pretreatment of the electrodes. The potentials at which the oxidation/reduction process occur, ensure that it is associated with Mg deposition/dissolution processes only and not with Li deposition. The latter process starts at approximately 1 V below Mg (see the inset to Figure 4).
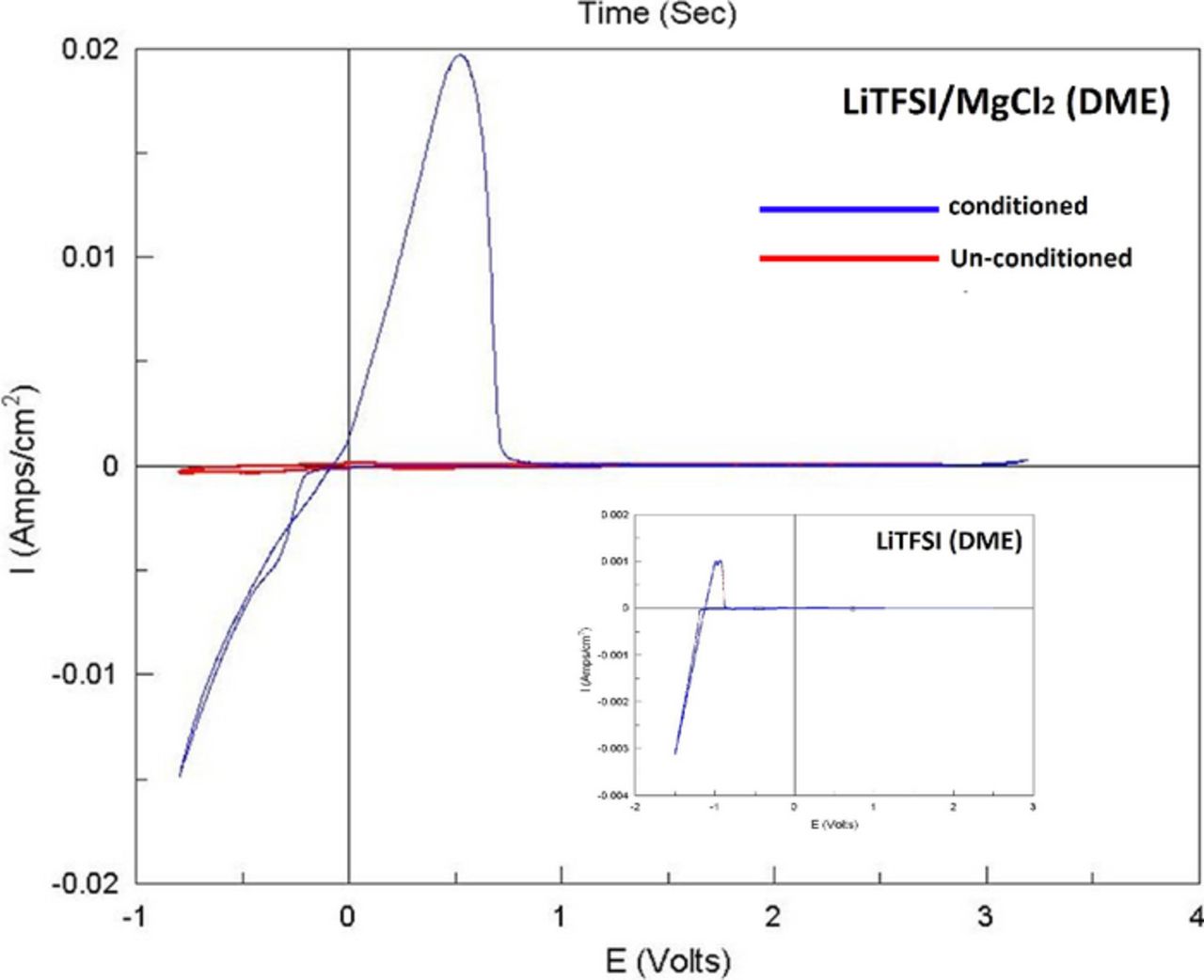
Figure 4. (a) Comparison between "conditioned" (blue curve) and untreated (red curve) LiTFSI 0.5 M + MgCl2 0.125 M in DME solutions, via their CV response with Pt WE (Mg foils RE and CE). The "conditioning" process included galvanostatic cycling of the cell until the overpotential for Mg deposition decreased below 0.2 V. The inset shows a CV depicting Li deposition/dissolution (on Pt WE), measured with a LiTFSI 0.5 M (DME) solution.
It is important to note that the conditioning processes for both MgTFSI2/MgCl2 and LiTFSI/MgCl2 solutions is mostly effective via a passage of a small amount of charge (deserves optimization) during prolonged cycling. If these solutions are pre-treated by cycling with a passage of too big amount of charge per cycle, they become contaminated by small floating Mg particles, formed due to dendritic Mg deposition processes (in contaminated solutions).
We explain the gradual decrease of Mg deposition over-potential during the conditioning (cycling) processes as follows: The pristine solutions contain unavoidably reactive contaminants such as trace water, oxygen, protic species etc., which are obviously reduced on the electrodes during cathodic polarization. For these specific solutions comprising DME, MgCl2 and TFSI based salts (Mg or Li), the irreversible cathodic processes related to contaminants don't form insoluble surface films that fully block the electrodes surfaces for any Mg deposition. However, some precipitation of insoluble surface species obviously occurs in pristine solutions, which impedes Mg deposition but does not fully avoid it. As cycling proceeds, the contaminants disappear from the solutions but their reduction products do not accumulate on the electrodes surface. The formation of the initial Mg deposits which are formed only at certain spots on the electrode's surface (which is not fully bare) requires high over-potentials. However, the coverage of the electrodes' surface by Mg deposits during repeated cycling increases, as the solutions contain less and less surface reactive species, which is naturally reflected by the observed lowering of the cathodic over-potential upon cycling.
So far, it was impossible to identify exactly how the pretreatment process works, what contaminants it removes and via which reactions. Spectroscopic studies of electrodes surfaces and solutions (Raman, FTIR) could not be conclusive, probably due to the relatively low concentration of the reactive contaminants. We assume that they may include traces of moisture, oxygen and HTFSI. We emphasize the uniqueness of these TFSI based solutions, in the fact that cathodic processes in them do not form surface films that block Mg deposition, as is the case with all other non-aqueous solutions containing conventional Mg salts. This uniqueness relates to the relative inertness of the main reactive components: DME and TFSI anions toward Mg surfaces and solvation properties related to DME/MgCl2 + MgTFSI2 or LiTFSI solutions that interfere with possible accumulation of the reduction by-products on the electrodes surfaces (Pt, Mg).
Based on the understanding above, pretreatment of DME/MgTFSI2/MgCl2 solutions can be achieved also chemically, by introducing to these solutions a trace reducing agent that can react effectively with traces of water, oxygen, and other protic moieties, without forming other detrimental contamimants. Di-butyl Mg seems to be a very suitable scavenger for pre-treating Mg salt solutions. It should react readily with water traces, oxygen and moieties like HTFSI, to form butane, Mg hydroxide (insoluble) Mg oxide (insoluble) and MgTFSI2, respectively. Such reactions in the solution bulk, avoid surface reactions of these reactive species that can passivate and block the electrodes. Indeed, addition of di-butyl-Mg to these solutions served as an effective pre-treatment that led to highly reversible Mg deposition without any adverse effect on the wide electrochemical window, as presented in Figure 5. As seen in this figure that shows steady state voltammograms of Pt electrodes with various DME/MgTFSI2/MgCl2/Bu2Mg solutions, addition of 250 ppm di-Butyl-Mg is an optimal amount for most cases to achieve good electrochemical performance. We have made sure that addition of 250 ppm does not substantiate di-butyl-Mg as the active species (as it is known that organo-magnesium species behave as active electrolytes in ether solutions2,3,25), by adding 250 ppm of Bu2Mg to LiTFSI/DME solutions that do not contain any Mg ions, verifying that no Mg related processes occur with such a concentration of Bu2Mg (inset to Fig. 5).
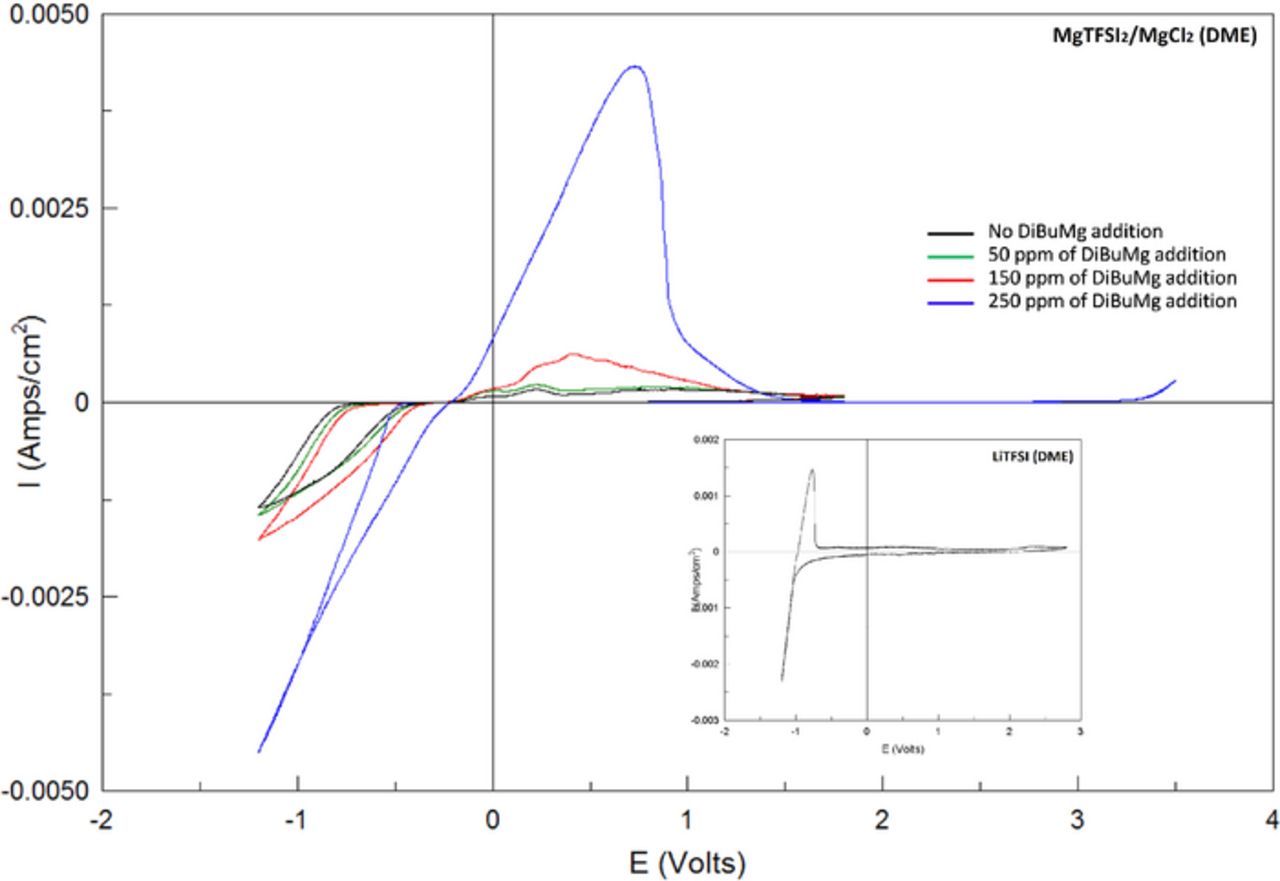
Figure 5. Cyclic voltammograms of a Pt electrodes in MgTFSI2 0.25 M + MgCl2 0.5 M (DME) solution with varying amounts of Bu2Mg. Mg foils used as RE and CE, scanned at 25 mV/sec. The inset shows a cyclic voltammogram of a Pt WE with a LiTFSI 0.5 M solution in DME with 250 ppm of Bu2Mg, in order to confirm that at this concentration, the latter moiety does not affect the electrochemical response.
Macro reversibility measurements, with preconditioned MgTFSI2/MgCl2 and LiTFSI/MgCl2 (DME) solutions were carried out. Mg was deposited with a charge of 0.5 coulomb per cm2, followed by galvanostaticaly cycling 20% of it (i.e. at 20% DOD) at 1 mA/cm2. Average cycling efficiencies in excess of 94% (MgTFSI2/MgCl2) and 96% (LiTFSI/MgCl2) respectively were obtained for these solutions after 100 cycles (see the formula in the Experimental section). Typical general voltage response of these experiments (left charts) plus selected voltage profiles in details (right charts) are presented in Figure 6 as indicated. Evidently, something is happening to the systems (the explored solutions and electrodes) during prolonged cycling, beyond the reversible Mg deposition and dissolution. The evolution of increasing over-potentials vs. time is clearly seen with both solutions. Possible major changes in the solutions chemistry, due to their repeated exposure to reactive Mg deposits, had been ruled out by ICP and Raman spectroscopic measurements. Thus, the gradual changes in the voltage profiles observed should be mainly attributed to surface chemistry and morphology aspects of the magnesium anodes. SEM micrographs obtained from electrodes after different stages of cycling showed virtually no morphological changes throughout the entire experiments. In any case, morphological changes during cycling usually results in ever increasing surface disorder and corrugation. This alone would lead to higher "real" surface area, which is expected to be reflected by smaller over-potentials. The most rational origin of this behavior is associated with continuous changes in the surface chemistry. If some irreversible reduction processes lead to accumulation of solid precipitates on the electrodes' surface during cycling, this can result is increasing overpotentials for deposition and dissolution. Such a scenario can be realized only if the alleged precipitate is permeable for magnesium ions. (The high cycling efficiency rules out the possibility of high permeability for other species.) Interestingly, the overpotential extent for the electrochemical processes is not symmetrical, but it does not show a coherent trend. These results demonstrate that the solution imparts a very unique electrochemical behavior with respect to the electrochemical magnesium deposition-dissolution processes. This behavior is strikingly different from that found for all aprotic, organic-solvents solutions with non-organometallic salts (provided that the solutions were extensively pure and dry). Backed by experimental work and comprehensive literature survey, the working hypothesis formulated is that magnesium anodes behave electrochemically reversible only under conditions when the system is of a metal-electrolyte interface type. That is to say, for a magnesium anode to be viable, it must possess no surface films at all. This is in downright contrast to lithium electrodes, which function always, and only, under conditions of stable SEI- shielded electrode, namely, solid electrolyte interphase. The reason for this is that except for very few cases (e.g: Chevrel phase Mo6S82,26 and V2O513,14,26), magnesium ions have very limited, or no mobility in the solid state (at, or near RT), a mandatory property for SEI layers.
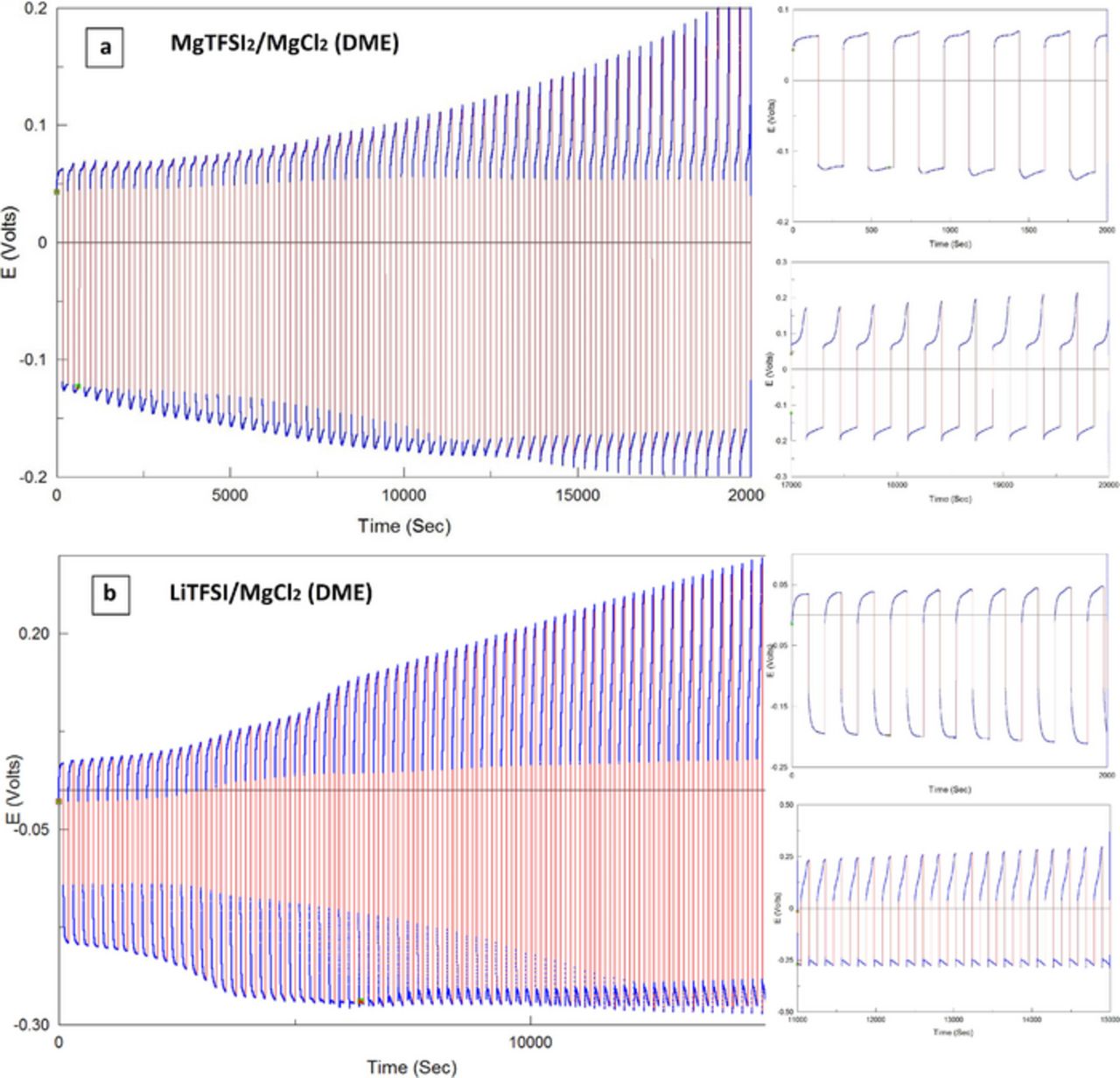
Figure 6. (a) Macro reversibility measurements for conditioned MgTFSI2 0.25 M + MgCl2 0.5 M/DME and LiTFSI 0.5 M + MgCl2 0.125 M/DME solutions. Certain amounts of magnesium were deposited on Pt electrodes (corresponding to 0.5 Coulombs per cm2). These Mg (on Pt) electrodes thus formed were cycled gavanostatically 100 times at 20% depth of discharge (around 0.1 Coulombs per cm2) and at 1 mA/cm2. Voltage profiles are presented (as indicated). The right charts show selected zoomed voltage profiles at the beginning and end of these experiments. Mg foils served as CE and RE.
To push the envelope further, we carried out similar macro cycling efficiency tests, with added "hold at OCV" periods of varying times. This kind of cycling schedule adds pressure on the system and allows time for the formation of surface films or corrosion. As can be seen in Figure 7, these OCV-hold periods seemed to have no impact on the cycling efficiency: the cycling curve resumes virtually from the point it was left prior to OCV-hold. The OCV value stabilizes at close to 0.0 V vs. Mg both for 1h and 24h hold periods. We conclude from this set of experiments that even relatively long immersion times do not result (at least not in a significant way) in Mg passivation or corrosion. Obviously, careful surface chemical analysis must augment these observations. However, so far, these results reflect the unique system stability and the prospects for cells showing high cycling life, low self-discharge and long shelf life. Nonetheless, we still believe that the TFSI anions play a critical role on the overall deposition reaction by either being reduced on the electrode around the magnesium deposition potential, or by reacting with the fresh, reactive magnesium deposit. In either case, the reaction products apparently do not lead to passivation, and are presumably, at least to some extent, soluble in the solution. The voltage profiles presented in Fig. 7 show, like the ones in Fig. 6, increasing deposition and oxidation over-potentials as a function of cycle number. Apparently, under the experiments conditions, this slight increase in over voltage that obviously reflects a gradual increase in impedance upon cycling is only a function of cycle number and not of time. The mechanisms underlying this increase in impedance are not understood yet. Hoverer, this kind of phenomenon is consistent with SEI-covered type situation, in which the SEI thickening with increasing cycle number leads to increased interfacial impedance. If this is the case, this is the first instance that a functioning reversible Mg anode is controlled by an SEI layer. Such a hypothesis needs thorough verification and characterization. It is, however possible that in the case that magnesium electrodes in such electrolyte solutions are indeed covered by some layer, this interphase does not behave as a classical SEI layer, but more like a selectively permeable membrane. The difference is, of course, small, but not merely semantic. The surface species that are probably formed on magnesium in these solutions allow a relatively rapid mobility for magnesium ions (monomeric or dimeric complexes, or just complexes with DME), but poor permeability for the TFSI ions (or ion-pairs, aggregates, etc.). Assuming that the conditioning processes clean out the solutions from reactive contaminants, the only candidate for surface reactions with Mg metal is the TFSI anions. We have previously explored in depth the surface chemistry of LiTFSI solutions in connection with Li ion batteries.27–30 The possible reduction processes of these anions are discussed in the next section. However, as we demonstrated several times before, the reduction processes of both solvents and salt anions depend very strongly on the identity of the cation. While TFSI anions reduction in the presence of Li ions can definitely form SEI type, highly passivating surface films,31 in the present case the driving force for TFSI reduction is much smaller, due to the higher potentials relevant. Thereby, TFSI anions reduction on Mg metal surface may not form uniformly cohesive and adhesive surface species that converge to SEI type (blocking for Mg ions) surface films. The permeability of Mg ions through surface structures that may be formed herein can probably be strongly assisted by the presence of chloride anions. The latter species are known to stabilize Mg ions in ether solutions. Their possible specific adsorption to Mg surfaces can further facilitate approach of Mg cations to the reductive electrode surface and help avoid formation of uniform SEI type surface films. In order to fully substantiate the assumptions above, further Mg surface studies in these solutions using spectro-electrochemical means are in progress.
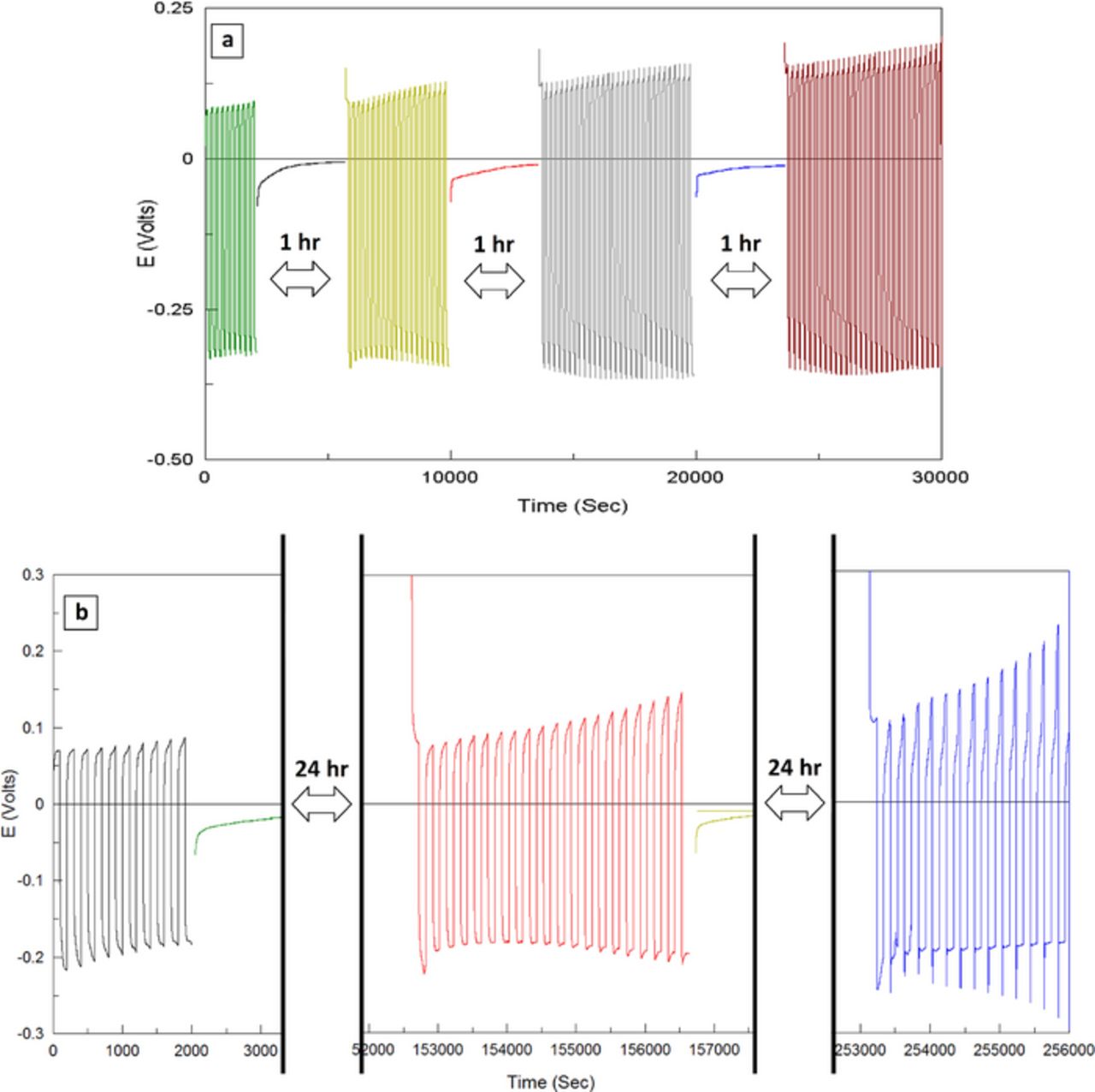
Figure 7. Macro reversibility cycling-hold measurements for conditioned MgTFSI2 0.25 M + MgCl2 0.5 M/DME solutions. Certain amounts of magnesium were deposited on Pt electrodes (corresponding to 0.5 Coulombs per cm2). These Mg (on Pt) electrodes thus formed were cycled gavanostatically at 20% depth of discharge (around 0.1 Coulombs per cm2) and at 1 mA/cm2. Voltage profiles are presented (as indicated). Mg foils served as CE and RE. (a) The electrodes were held for 1 hr at OCV after each cycling period. (b) The electrodes here held for 24 hrs at OCV after each cycling period.
Morphology and surface chemistry
One of the important factors for "good" electrolyte solutions for Mg metal based batteries is their ability to upkeep uniform, dendrite-free metallic deposits during charging. At the same token, the discharge process should be carried out in a homogeneous way, leaving flat, pinhole-free layers. To examine the electrodeposited Mg morphology obtained in these solutions, versus the deposition current density, about 1 micrometer thick layer (calculated, based on charge density and metal density) of Mg was deposited from previously conditioned solutions of MgTFSI2(0.25 M)+MgCl2(0.5 M) and LiTFSI(0.5 M)+MgCl2(0.125 M) in DME, on Pt electrodes at various current densities ranging from 1 to 5 mA/cm2. The Scanning electron micrographs of the deposited Mg are seen in Figure 8. The magnesium deposits in both solutions exhibits sharp, crystalline particles associated with "good" metal deposition, as observed in previous studies with organometallic complexes based electrolyte solutions.4,32 Current density influences the morphology of the Mg deposits. At higher current densities the deposition seems more edgy and rough though equally distributed. No dendrites were observed at current densities of up to 5 mA/cm2, even after prolonged cycling. EDS measurements have confirmed the presence of Mg on the Pt electrode (Fig. 8f), no Mg was observed on Pt electrode which underwent a full deposition and dissolution cycles, confirming that the reduction currents are in-fact linked only to Mg deposition. It is fair to say however, that we cannot at this point rule out completely co-deposition process of both Li and Mg in LiTFSI/MgCl2 solutions since the EDS sensitivity for Li is very poor. Nonetheless, Co-deposition process is unlikely since as can be seen from Figure 4b (inset), Li deposition occurs at a much lower potential (1 V) than Mg. Detection of sulfur with EDS is our best candidate to confirm formation of by-products resulting from TFSI and Mg reaction, since fluorine, carbon and nitrogen moieties have poor detection limits by this technique. Only minor amount of sulfur was detected, conveying that the by-product layer is relatively thin. In contrast to the crystalline Mg deposition obtained from solutions containing MgCl2, in pure MgTFSI2 (DME) solutions no crystalline Mg formation could be identified (Figure 8e). The deposits seem amorphous and more organic in nature suggesting formation of by-products during reduction. Clearly, MgCl2 is a major factor for obtaining normal Mg deposition conditions. As suggested above, possible adsorption of chloride anions on Mg surfaces may inhibit an effective reduction of TFSI anions in these systems. Scheme 1 presents TFSI anions reduction reactions related to non-aqueous Li electrochemistry, composed based on previous studies. It was proven that TFSI anions reduction on graphite surfaces in the presence of Li ions (occurring at potentials <1 V vs Li) form passivating SEI type surface films, composed of the end products of the reactions suggested in Scheme 1.

Figure 8. SEM images of a 1 micron thick (calculated) Mg deposits on Pt plated glass slides. The Mg was deposited from: (a) MgTFSI2 0.25 M + MgCl2 0.5 M/ DME solution at 1 mA/cm2, (b) MgTFSI2 0.25 M + MgCl2 0.5 M/DME solution at 5 mA/cm2, (c) LiTFSI 0.5 M + MgCl2 0.125M/DME solution at 1 mA/cm2, (d) LiTFSI 0.5 M + MgCl2 0.125M/DME solution at 4 mA/cm2, (e) MgTFSI2 0.5 M/DME solution at 1 mA/cm2. (f) Typical EDS spectra for deposited Mg on Pt obtained from MgTFSI2 or LiTFSI/MgCl2 /DME solutions.
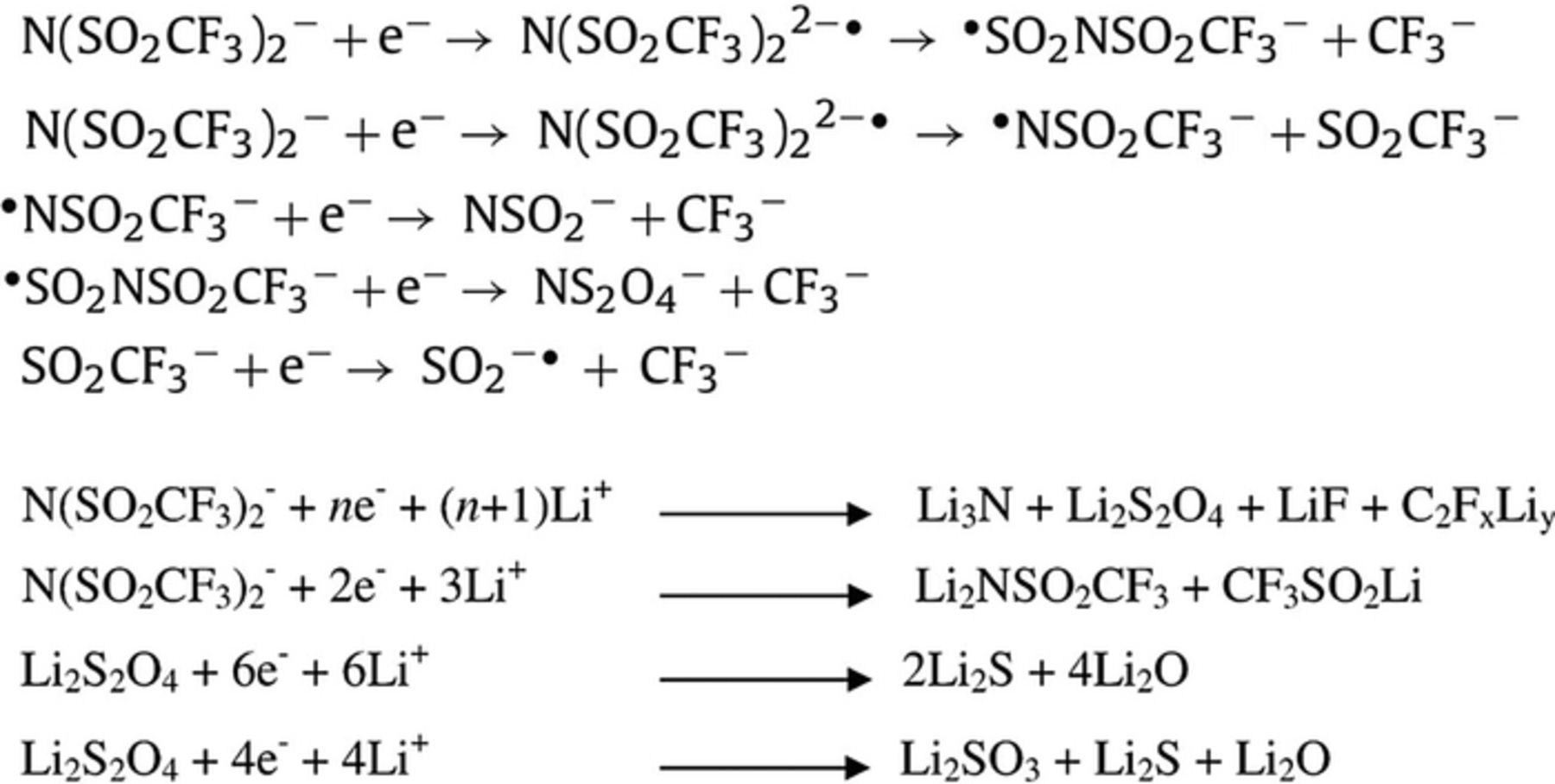
Scheme 1. Possible reduction reactions of LiTFSI in non-aqueous solutions, based on Refs. 24 and 25.
XPS measurements were carried out to detect any surface film formation on the freshly deposited Mg in the TFSI based solutions discussed herein (Fig. 9). XPS measured from similar magnesium deposits have shown the expected Mg 2p, Mg 2s and the Auger multiplet' peaks, but only relatively minor Cl, S and F related peaks. Oxygen and carbon are expected to be detected in large concentrations irrespective to any chemical reactions. The XPS results further solidify the notion that no stable passivation layers are formed on the surface, what allows the continuation of the Mg deposition/dissolution process at relatively low over-potentials. We can definitely claim that if some reactions between TFSI ions and Mg metal are possible, they are completely different than those occurring in Li salt solutions at lower potentials (≪ 0 V vs. Mg).
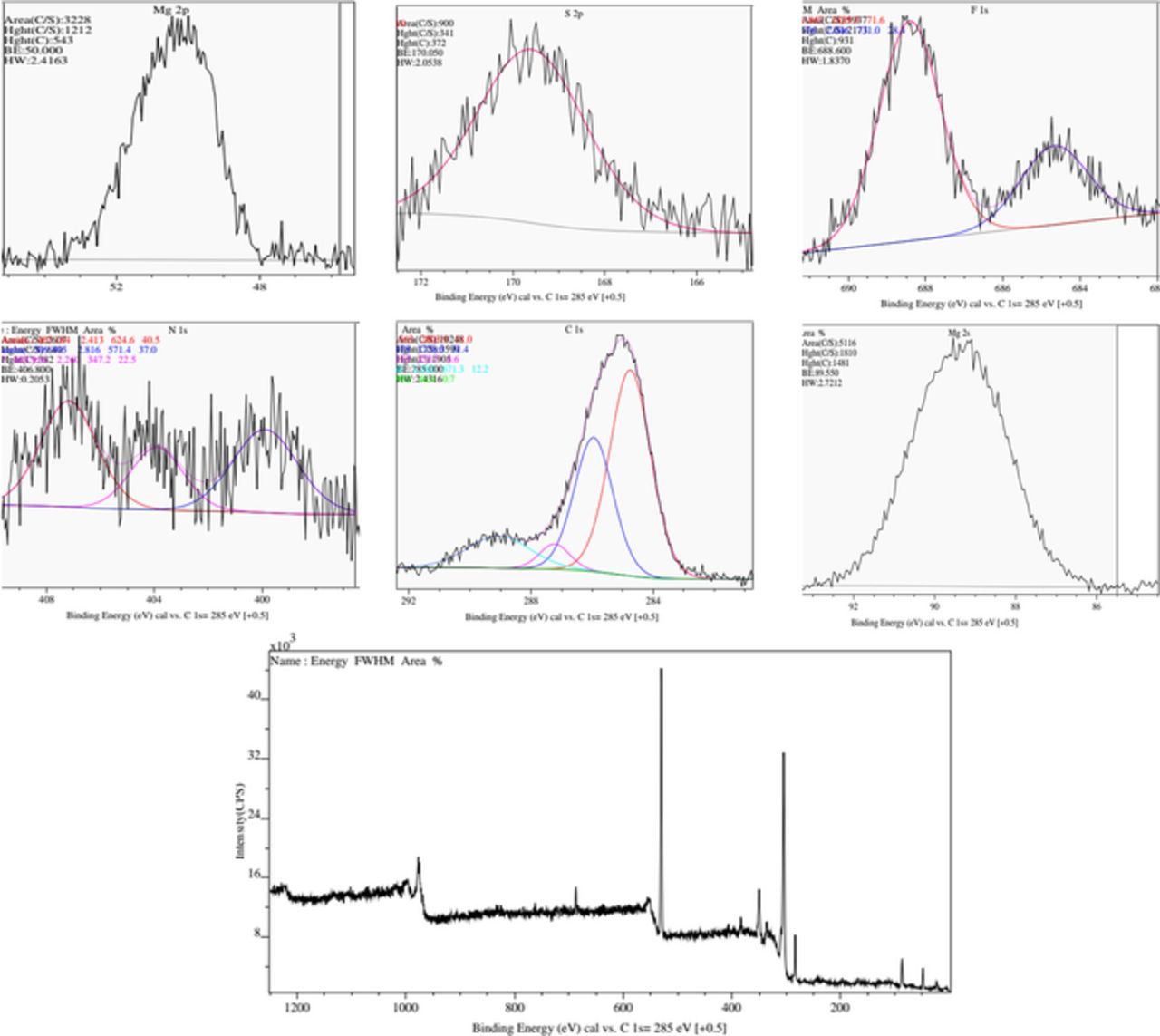
Figure 9. XPS spectra measured with Mg deposits from a LiTFSI 0.5 M + MgCl2 0.125 M/ DME solution. For these measurements, Mg was deposited on Ni plated glass slides. Six high resolution spectra with Shirley baseline are presented as indicated for Mg 2p, S 2p, F 1s, N 1s, C 1s, and Mg 2s. Peak fitting is presented for F 1s, N 1s and C 1s. Bottom: a typical overall survey spectrum of the Mg sample. No other elements were detected on these samples surface.
Intercalation into Mo6S8/V205
After we demonstrated the compatibility of DME/MgTFSI2/MgCl2 solutions with reversible Mg anodes, it is crucially important to examine suitability with relevant cathodes. These solutions should a) allow reversible reactions (most preferred intercalation/de-intercalation) of Mg ions with cathode materials and b) avoid side reactions with such cathode materials. The first obvious step is to verify compatibility with a cathode material known to intercalate Mg ions reversibly at fast rates. There is only one such material at the moment, Chevrel phases of the MgxMo6X8 type (0 < x < 2, X = S, Se). Indeed, Mg ions can be reversibly intercalated into Chavrel phase cathodes, for many cycles and without any capacity fading, from conditioned MgTFSI2/MgCl2 (DME) solutions (Fig. 10a) with a specific capacity approaching 80 mAh/gr. Since the experiments related to Figure 10a presented herein are only preliminary, we assume that higher specific capacities can be obtained with Chevrel phase cathodes in these solutions (a theoretical capacity of 120 mAh/g). Attempts to realize the possibility of reversible Mg intercalation into other hosts from these solutions are being made in parallel studies.
Turning to DME/LiTFSI/MgCl2 solutions, in contrast to the difficulties in Mg ions intercalation into any transition metal oxide hosts, the Li ions in these solutions can readily and reversibly intercalate into such hosts provided that their red-ox activity falls within their electrochemical window (<3.1 V vs. Mg).
Excellent examples are V2O5 cathodes, whose red-ox potentials for the following processes: V2O5 + 0.5 Li+ + 0.5 e− → Li0.5V2O5; Li0.5V2O5 + 0.5 Li+ + 0.5 e− → LiV2O5 occur in the range of 3–4 vs. Li = 2–3 V vs. Mg.33 Figure 10b shows typical steady state voltammograms of composite V2O5 electrodes in a pretreated DME/LiTFSI/MgCl2 solution, reflecting fully reversible behavior at 100% efficiency. The results presented in Figures 10a and 10b are of a preliminary nature, as interactions with cathode materials are beyond the scope of the studies reported herein.
Of course these solutions cannot be implemented in starved cell configurations. However, in systems which allow high enough solution volume to electrodes surface ratio such systems would benefit from the possibility of using a high-capacity, dendrite-free reversible Mg anodes coupled with a fast Li+ intercalation/de-intercalation process on the cathode side, unhindered by the slow insertion kinetics of Mg2+ ions.
Conclusions
In this work we have evaluated the electrochemical performance of TFSI based electrolyte solutions. We showed that solutions comprising MgTFSI2 in DME have poor electrochemical performance, in terms of reversible magnesium deposition. Addition of MgCl2 to these solutions significantly improves their electrochemical performance and enables reversible Mg deposition. An obvious role of the chloride anions in solutions is stabilization of Mg cations. They may also have an important role in facilitating the approach of Mg ions to the electrode surface and inhibition of passivation phenomena. We have conjectured that the electrochemical performance of these TFSI based electrolyte solutions is governed by their purity level, which can be considerably improved by electrochemical and chemical (addition of Bu2Mg) treatments. It was possible to obtain reversible Mg deposition approaching very high cycling efficiencies with purified DME/MgTFSI2/MgCl2 solutions. Using LiTFSI instead of MgTFSI2 in these solutions enabled us to reach Mg deposition/dissolution cycling efficiency reaching 100%, due to the higher level of purity of the Li salt, without any interference from any Li ions activity. The Mg deposits from MgTFSI2 or LiTFSI/MgCl2/DME solutions were found to be uniform and crystalline in nature, confirming to a standard Mg deposition. The electrochemical window of these solutions is >3 V, reflecting the anodic stability of ether solvents, what may allow the use of attractive high capacity cathode materials. The MgTFSI2/MgCl2/DME solutions were found to be fully compatible with Chevrel phase cathodes. However, possible Mg ions insertion into hosts such as transition metal oxides is very questionable. In turn, lithiation of V2O5 electrodes in LiTFSI/MgCl2/DME solutions is fully reversible. Macro-reversibility measurements showed that the over-potentials of repeated Mg deposition and dissolution processes slightly and gradually increase during prolonged cycling. We showed that this steady increase in the over-potentials is related to the cycle number rather than the retention time. This phenomenon is most likely related to accumulation of precipitates on the electrode, probably due to some surface reactions related to the TFSI anions. The surface species thus formed do not converge to SEI type surface films, as their precipitation does not avoid smooth Mg deposition/dissolution processes. These surface processes have a slight impedance effect, which is reflected by the observed increase in the over-potential of Mg deposition/dissolution. It can be stated that the performance of MgTFSI2 or LiTFSI/MgCl2/DME solutions shows promising signs and have a good potential to be used in rechargeable Mg batteries. Main questions which are yet un-resolved and are being investigated, relate to the true stability of the TFSI anions, the possible interfacial role of chloride ions and compatibility of these solutions with transition metal oxide cathode materials.
Acknowledgment
Partial support for this work was obtained by the Israel Science Foundation, ISF in the framework of the INREP project.