
Abstract
 , which can be synthesized either via an ionothermal or solid-state route, stands as a possible alternative to
, which can be synthesized either via an ionothermal or solid-state route, stands as a possible alternative to  for the next generation of Li-ion batteries. Here we demonstrate a different route to prepare this material. It consists of (i) combining stoichiometric amounts of
for the next generation of Li-ion batteries. Here we demonstrate a different route to prepare this material. It consists of (i) combining stoichiometric amounts of  and LiF in a powdered polymeric media, which has a low melting point and remains stable up to
and LiF in a powdered polymeric media, which has a low melting point and remains stable up to  , and (ii) recovering and purifying the reacted powder by washing it in organic solvent. This method offers advantages in terms of both cost and kinetics while providing powders that have similar performances vs Li.
, and (ii) recovering and purifying the reacted powder by washing it in organic solvent. This method offers advantages in terms of both cost and kinetics while providing powders that have similar performances vs Li.
Export citation and abstract BibTeX RIS
Ionothermal synthesis, i.e., the use of ionic liquids as a synthetic medium, has been used recently to prepare a  fluorosulfate,1 which is able to reversibly intercalate/deintercalate Li at 3.6 V vs Li, thus making this material a serious contender of
fluorosulfate,1 which is able to reversibly intercalate/deintercalate Li at 3.6 V vs Li, thus making this material a serious contender of  2 for electric vehicle applications. Although fluorosulfates are made from low cost and abundant chemical elements, their synthetic process relies on the use of costly ionic liquids and the economy of the process depends on how efficiently it can be recovered and reused multiple times. With this in mind, we recently developed an alternative low temperature solid-state synthetic approach to
2 for electric vehicle applications. Although fluorosulfates are made from low cost and abundant chemical elements, their synthetic process relies on the use of costly ionic liquids and the economy of the process depends on how efficiently it can be recovered and reused multiple times. With this in mind, we recently developed an alternative low temperature solid-state synthetic approach to  .3 It simply consists in reacting
.3 It simply consists in reacting  and LiF (in slight stoichiometric excess) in a dry and closed environment at
and LiF (in slight stoichiometric excess) in a dry and closed environment at  for 48 h. Despite the obvious cost advantages, this new solid-state synthetic route has two distinct disadvantages compared to ionothermal synthesis: (i) The reaction mechanism was slower than when performed in ionic liquids, leading to longer reaction times (48 h). (ii) The solid-state process requires operating with an excess of LiF; therefore, the resulting powders are contaminated by small amounts of LiF. This electrochemically inactive impurity acts as a dead weight in practice, lowering the electrode capacity. To date, the best materials thus obtained had 0.15 mol excess of LiF, required 72 h of reaction times, and delivered reversible capacities of 0.8 Li per formula unit. In contrast, ionothermal synthesis has led to single-phased materials coming close to 0.9 Li in reversible capacity, which can be made in 24 h by reacting stoichiometric amounts of precursors.
for 48 h. Despite the obvious cost advantages, this new solid-state synthetic route has two distinct disadvantages compared to ionothermal synthesis: (i) The reaction mechanism was slower than when performed in ionic liquids, leading to longer reaction times (48 h). (ii) The solid-state process requires operating with an excess of LiF; therefore, the resulting powders are contaminated by small amounts of LiF. This electrochemically inactive impurity acts as a dead weight in practice, lowering the electrode capacity. To date, the best materials thus obtained had 0.15 mol excess of LiF, required 72 h of reaction times, and delivered reversible capacities of 0.8 Li per formula unit. In contrast, ionothermal synthesis has led to single-phased materials coming close to 0.9 Li in reversible capacity, which can be made in 24 h by reacting stoichiometric amounts of precursors.
Both processes present great attributes as they both occur at low temperatures, but neither the ionothermal process nor the ceramic processes were ideal because of costs and kinetics issues, respectively. Thus, we searched for a reaction medium that could gather the benefits offered by both the ceramic and ionothermal processes. In practice, we searched for a synthetic medium, which could be handled as a powder at room temperature, become a liquid at moderate temperatures, and then have an excellent thermal stability well above  . Poly(ethylene glycol) (PEG) (depending on its
. Poly(ethylene glycol) (PEG) (depending on its  ) offers such a possibility as it is solid at room temperature, melts at
) offers such a possibility as it is solid at room temperature, melts at  , and is stable up to temperatures as high as
, and is stable up to temperatures as high as  . Besides, several groups have recently used glycols in solvothermal reactions to make related polyanionic compounds.4–6
. Besides, several groups have recently used glycols in solvothermal reactions to make related polyanionic compounds.4–6
Experimental
Previous studies clearly indicate that the preparation of 3d-metal fluorosulfates, having the tavorite  or derived tavorite structure
or derived tavorite structure  , can be made via a topotactic reaction involving the removal of one water molecule from the precursor phase
, can be made via a topotactic reaction involving the removal of one water molecule from the precursor phase  and the ingress of LiF. The success in conducting such a reaction depends on the kinetics of both the water removal and the LiF ingress with the formation of an inert secondary phase
and the ingress of LiF. The success in conducting such a reaction depends on the kinetics of both the water removal and the LiF ingress with the formation of an inert secondary phase  when the reaction kinetics is unfit, i.e., when the water is removed too quickly.1 Hydrophobic ionic liquids such as 1-ethyl-3-methylimidazolium bis (trifluoromethanesulfonyl imide) (EMI-TFSI, Solvoionic Inc.) were shown, owing to their encapsulating role, to delay the removal of water, thereby enabling the preparation of pure fluorosulfate powders. An alternative way to control water departure was provided by operating in a confined environment (Parr bombs) as recently reported by our group.3
when the reaction kinetics is unfit, i.e., when the water is removed too quickly.1 Hydrophobic ionic liquids such as 1-ethyl-3-methylimidazolium bis (trifluoromethanesulfonyl imide) (EMI-TFSI, Solvoionic Inc.) were shown, owing to their encapsulating role, to delay the removal of water, thereby enabling the preparation of pure fluorosulfate powders. An alternative way to control water departure was provided by operating in a confined environment (Parr bombs) as recently reported by our group.3
In line with these recent findings, it was conceivable that other solvents could have this same effect on the dehydration rates while providing cost, handling, and stability advantages. Hence, we have embarked on a survey of reaction media, mainly polymers, which are solid at room temperature, liquid at elevated temperatures, and have viscosities and thermal stabilities close to those of ionic liquids at  . Our proof-of-concept studies are based on the studied PEG-type polymers. Such polymers, which melt at temperatures near
. Our proof-of-concept studies are based on the studied PEG-type polymers. Such polymers, which melt at temperatures near  , offer, depending on their
, offer, depending on their  , high temperature stability in their liquid state. For instance, the thermal stability of PEGs (Fig. 1) changes from
, high temperature stability in their liquid state. For instance, the thermal stability of PEGs (Fig. 1) changes from 
 to
to 
 ) and increases even more
) and increases even more  , well above the synthesis temperatures of
, well above the synthesis temperatures of 
 , for poly(ethylene oxide)s (PEOs) having
, for poly(ethylene oxide)s (PEOs) having  approaching 5,000,000 a.u.
approaching 5,000,000 a.u.
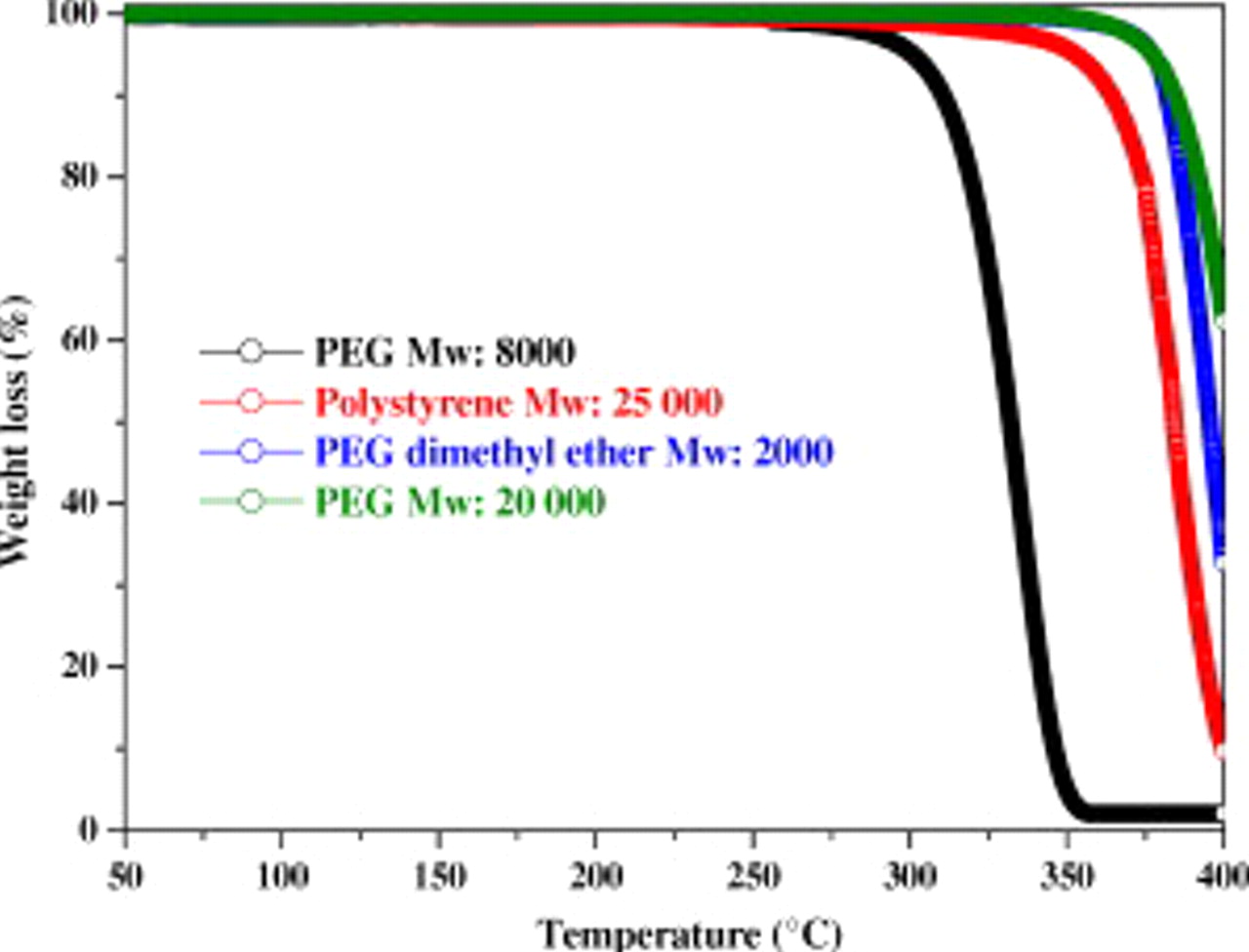
Figure 1. Thermogravimetric analysis of the PEG  polymer (green solid line), PEG dimethyl ether
polymer (green solid line), PEG dimethyl ether  (blue solid line), polystyrene
(blue solid line), polystyrene  (red line), and PEG
(red line), and PEG  (black solid line) using Ar as gas vector and a heating rate of
(black solid line) using Ar as gas vector and a heating rate of  showing the effect of
showing the effect of  on the thermal stability.
on the thermal stability.
A typical synthetic protocol for the preparation of  in a polymeric media involves several steps consisting in (i) ballmilling for 10 min in an SPEX 800 miller, 0.85 g of
in a polymeric media involves several steps consisting in (i) ballmilling for 10 min in an SPEX 800 miller, 0.85 g of  , prepared as previously described,3 with a stoichiometric amount of LiF (0.13 g); (ii) pouring this mixture, loosely mixed with ≈3 g of PEG
, prepared as previously described,3 with a stoichiometric amount of LiF (0.13 g); (ii) pouring this mixture, loosely mixed with ≈3 g of PEG  , into a poly(tetrafluoroethylene) (PTFE)-lined steel bomb, which was closed under argon before being placed into an oven; and (iii) increasing the oven temperature to
, into a poly(tetrafluoroethylene) (PTFE)-lined steel bomb, which was closed under argon before being placed into an oven; and (iii) increasing the oven temperature to  over 2 h, maintained at that temperature for 24 h, and cooling the powder to ambient temperature by switching off the oven. In addition to PEG, both triethylene glycol and monoethylene glycol solvent systems were tested using both thermal and microwave heatings but failed to produce the material.
over 2 h, maintained at that temperature for 24 h, and cooling the powder to ambient temperature by switching off the oven. In addition to PEG, both triethylene glycol and monoethylene glycol solvent systems were tested using both thermal and microwave heatings but failed to produce the material.
The recovered solid residue was then washed several times with ethyl acetate, or even better with methyl formate, to remove the polymer matrix; then it was oven-dried under air at  before being characterized by X-ray diffraction.
before being characterized by X-ray diffraction.
Results and Discussion
The XRD patterns (Fig. 2a, 2b and 2c), recorded on a Bruker D8 Advance diffractometer ( radiation,
radiation,  ), indicate the presence of the single-phase
), indicate the presence of the single-phase  as all the peaks were entirely indexed in the space group
as all the peaks were entirely indexed in the space group  . The lattice parameters
. The lattice parameters  ,
,  ,
,  ,
,  ,
,  ,
,  , and
, and  obtained from full pattern matching refinements via the Fullprof program7 agree well with our previous article. The scanning electron microscope (SEM) images of the powders, taken with a field-emission gun (FEI Quanta F200P), revealed nicely crystallized
obtained from full pattern matching refinements via the Fullprof program7 agree well with our previous article. The scanning electron microscope (SEM) images of the powders, taken with a field-emission gun (FEI Quanta F200P), revealed nicely crystallized  cubic particles (Fig. 2
cubic particles (Fig. 2
 ).
).
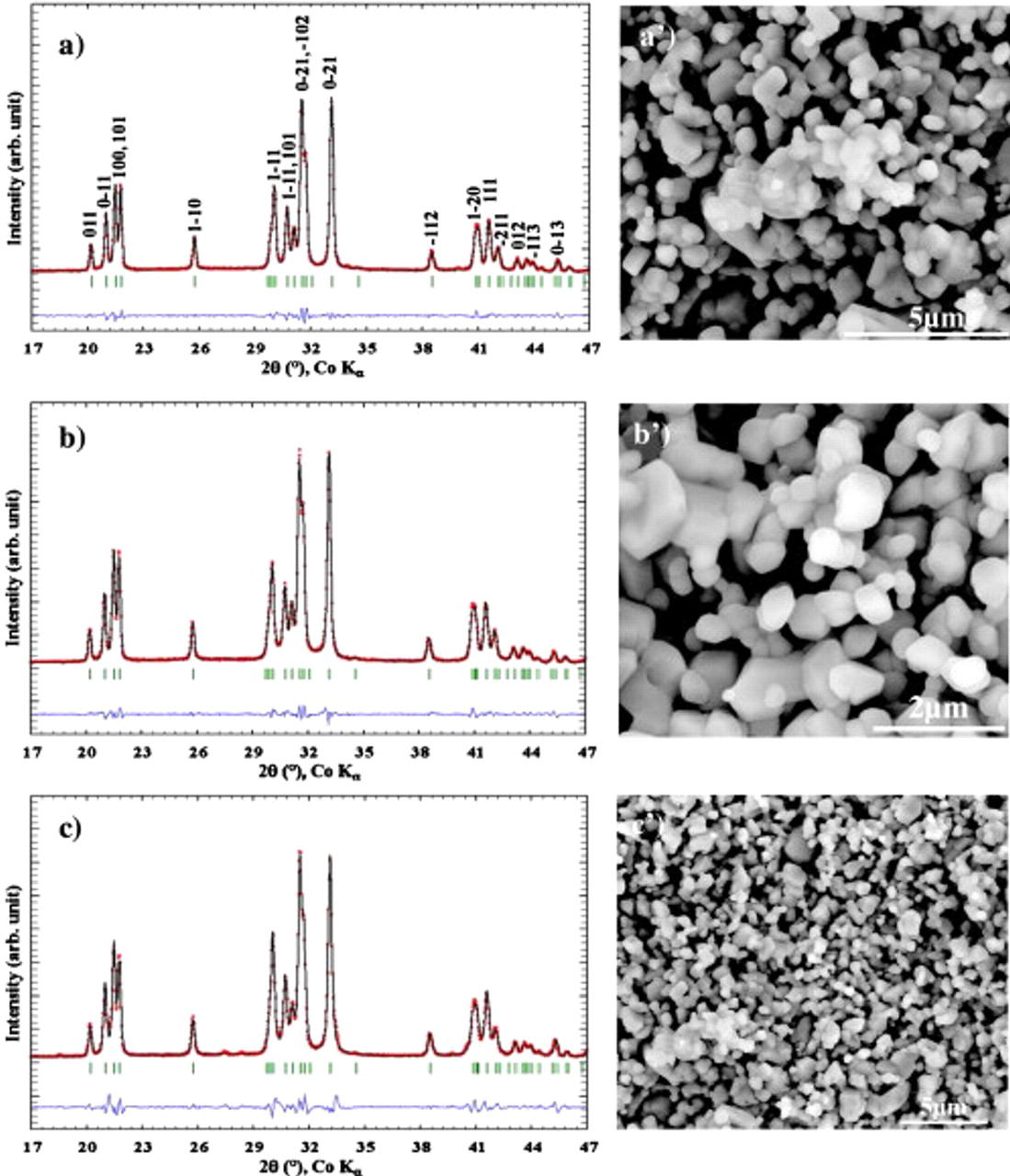
Figure 2. XRD patterns of the  material made using (a) PEG
material made using (a) PEG  polymer, (b) PEG dimethyl ether
polymer, (b) PEG dimethyl ether  , and (c) polystyrene
, and (c) polystyrene  and their corresponding SEM pictures
and their corresponding SEM pictures  ,
,  , and
, and  .
.
With this concept validated, it was then extended to various polymeric materials with various  as well as chemical nature. The first variable that we explored was the importance of the
as well as chemical nature. The first variable that we explored was the importance of the  of both PEG/PEO (PEG and PEO differ only in terms of
of both PEG/PEO (PEG and PEO differ only in terms of  ranges,
ranges,  and
and  ). As a consequence of PEO's higher
). As a consequence of PEO's higher  , the PEO polymers both have a higher viscosity and a melting point of
, the PEO polymers both have a higher viscosity and a melting point of  . Table I contains information on some of the polymers that we used, the reaction conditions, and the resulting phases. Using a similar synthetic protocol as described above, we succeeded in preparing a single-phase
. Table I contains information on some of the polymers that we used, the reaction conditions, and the resulting phases. Using a similar synthetic protocol as described above, we succeeded in preparing a single-phase  with PEO
with PEO  while our attempts to prepare a single-phased material with PEG
while our attempts to prepare a single-phased material with PEG  failed. Regardless of the thermal treatment utilized, the samples synthesized with PEG
failed. Regardless of the thermal treatment utilized, the samples synthesized with PEG  were always contaminated with large amounts (≈20%) of
were always contaminated with large amounts (≈20%) of  . This failure most likely results from the limited thermal stability of PEG 8000 (
. This failure most likely results from the limited thermal stability of PEG 8000 ( , onset see Fig. 1).
, onset see Fig. 1).
Table I. Respective names, formulas, and thermal properties of the polymers used to prepare the different fluorosulfate phases  with (
with ( and Na and
and Na and  , Co, and Ni) specifying the conditions of synthesis and the purity of the obtained materials.
, Co, and Ni) specifying the conditions of synthesis and the purity of the obtained materials.
Thermal decomposition of PEG polymers takes place at the terminal OH groups. To further test this thermal stability issue and the possible H-bond solvation explaining the failure of simple glycol, we tried low  polymers having both one terminal ether (PEG monoethyl ether
polymers having both one terminal ether (PEG monoethyl ether  ) or two terminal ether groups (PEG dimethyl ether
) or two terminal ether groups (PEG dimethyl ether  ), with decomposition temperatures greater than
), with decomposition temperatures greater than  . Both polymers led to single-phased
. Both polymers led to single-phased  samples with micrometric particle sizes. In an attempt to control the particle size, we decided to explore the use of block copolymers rather than single component polymer precursors. PEG-block copolymer-poly(propylene) oxide (PPO)-block copolymers PEG (PEG–PPO–PEG) have been widely reported8 to allow partition space at the nanometer level (synthesis of Fe nonoparticles and mesoporous materials). Generally, the block-copolymer acts as a surfactant to form some micelles, which act as nanoreactors to lead to the formation of nanomaterials. To test this concept, we first chose a PEG–PPO–PEG block copolymer with
samples with micrometric particle sizes. In an attempt to control the particle size, we decided to explore the use of block copolymers rather than single component polymer precursors. PEG-block copolymer-poly(propylene) oxide (PPO)-block copolymers PEG (PEG–PPO–PEG) have been widely reported8 to allow partition space at the nanometer level (synthesis of Fe nonoparticles and mesoporous materials). Generally, the block-copolymer acts as a surfactant to form some micelles, which act as nanoreactors to lead to the formation of nanomaterials. To test this concept, we first chose a PEG–PPO–PEG block copolymer with  and a decomposition temperature greater than
and a decomposition temperature greater than  . As was the case for the PEG- or PEO-based copolymers, we were able to prepare single-phased
. As was the case for the PEG- or PEO-based copolymers, we were able to prepare single-phased  within such a reacting medium as well; therefore, no drastic differences were noted in the particle size with powders directly grown in PEG reacting media. In contrast, the use of the block copolymer PEO–block-PEG (
within such a reacting medium as well; therefore, no drastic differences were noted in the particle size with powders directly grown in PEG reacting media. In contrast, the use of the block copolymer PEO–block-PEG ( PEO) failed whatever the reacting temperature, and we again ascribed this failure to its low thermal decomposition temperature
PEO) failed whatever the reacting temperature, and we again ascribed this failure to its low thermal decomposition temperature  .
.
To this point, we have focused on the thermal stability of the polymer solvent system for the successful synthesis of  while ignoring contributions associated with solvent polarity. To determine the role of solvent polarity, polystyrene (a nonpolar polymer) with
while ignoring contributions associated with solvent polarity. To determine the role of solvent polarity, polystyrene (a nonpolar polymer) with  ,
,  , and a thermal stability approaching
, and a thermal stability approaching  was used to perform the synthesis. Using a mixture of 0.85 g of
was used to perform the synthesis. Using a mixture of 0.85 g of  , 0.13 g of LiF, 3 g of polystyrene, and a reaction time of 24 h at
, 0.13 g of LiF, 3 g of polystyrene, and a reaction time of 24 h at  , we were able to prepare single-phased materials, further indicating that thermal stability rather than solvent properties is fundamental when selecting the proper polymers. This did not come as a total surprise as we have previously shown that the ionothermal synthesis of such a phase proceeded via a topotactic solid-state reaction. However, recovering the final product from a polystyrene mixture is not an easy task as it requires at least five consecutive washes with methyl formate as compared to only two washes when PEG polymers are used. The use of end-capped lower
, we were able to prepare single-phased materials, further indicating that thermal stability rather than solvent properties is fundamental when selecting the proper polymers. This did not come as a total surprise as we have previously shown that the ionothermal synthesis of such a phase proceeded via a topotactic solid-state reaction. However, recovering the final product from a polystyrene mixture is not an easy task as it requires at least five consecutive washes with methyl formate as compared to only two washes when PEG polymers are used. The use of end-capped lower  polymer may ease this polymer/powder separation to perform single-wash purification.
polymer may ease this polymer/powder separation to perform single-wash purification.
To further explore the opportunity provided by this synthetic protocol, we applied this approach to the synthesis of other fluorosulfates having either different 3d metals ( and
and  ) or alkali metals
) or alkali metals  . To conduct such synthesis, stoichiometric amounts of
. To conduct such synthesis, stoichiometric amounts of 
 and alkali fluorides AF
and alkali fluorides AF  were placed in a ballmilling cell with 1 cm diameter stainless steel balls; the cell was then closed under Ar. Next, around 2 g of PEG
were placed in a ballmilling cell with 1 cm diameter stainless steel balls; the cell was then closed under Ar. Next, around 2 g of PEG  was poured in the bottom of the Teflon-lined autoclave; the powder was placed in the middle and covered again with 2 g more of PEG. The PTFE-lined autoclave was closed under Ar and placed into an oven whose temperature was brought to
was poured in the bottom of the Teflon-lined autoclave; the powder was placed in the middle and covered again with 2 g more of PEG. The PTFE-lined autoclave was closed under Ar and placed into an oven whose temperature was brought to  for 24 h. Under such a procedure, single phases of
for 24 h. Under such a procedure, single phases of  ,
,  , and
, and  having lattice parameters as previously reported (Table II) were obtained.
having lattice parameters as previously reported (Table II) were obtained.
Table II. Crystallographic data of the different prepared phases of  with "
with " and Na" and "
and Na" and " , Co, and Ni."
, Co, and Ni."
| Name |
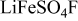
|
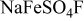
|
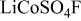
|
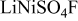
|
|---|---|---|---|---|
| Space group |
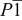
|
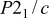
|
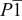
|
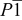
|
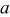 (Å) (Å) | 5.1747(3) | 6.679(3) | 5.1719(6) | 5.173(1) |
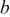 (Å) (Å) | 5.4943(3) | 8.7061(3) | 5.4192(6) | 5.421(4) |
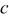 (Å) (Å) | 7.224(2) | 7.1912(2) | 7.1818(7) | 7.183(2) |
| α (Å) | 106.522(3) | 90 | 106.811(7) | 106.828(8) |
| β (Å) | 107.210(3) | 90 | 107.771(7) | 107.776(7) |
| γ (Å) | 97.791(3) | 113.517(2) | 97.975(5) | 97.923(8) |
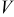 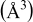
| 182.559(2) | 383.473(2) | 117.71(3) | 177.85(5) |
To check the electrochemical performances of the various batches of  powders prepared in various polymers,
powders prepared in various polymers,  Swagelok cells, using 1 M
Swagelok cells, using 1 M  electrolyte solution in (1:1) dimethyl carbonate–ethylene carbonate as electrolyte, were assembled in an argon-filled glove box with a glass fiber separator. A positive electrode mix was obtained by ballmilling
electrolyte solution in (1:1) dimethyl carbonate–ethylene carbonate as electrolyte, were assembled in an argon-filled glove box with a glass fiber separator. A positive electrode mix was obtained by ballmilling  with 20% w/w carbon black (SP) for 10 min and loadings of
with 20% w/w carbon black (SP) for 10 min and loadings of  of the active material were used. The counter electrodes were metallic Li. All single-phased
of the active material were used. The counter electrodes were metallic Li. All single-phased  samples were electrochemically active. Therefore, to be brief, we only report data for the sample made with the PEG
samples were electrochemically active. Therefore, to be brief, we only report data for the sample made with the PEG  . The voltage composition trace for a
. The voltage composition trace for a  cell cycled between 4.2 and 2.5 V at a C/20 rate (1 Li in 20 h) shows a sustained reversible electrochemical activity of 0.8
cell cycled between 4.2 and 2.5 V at a C/20 rate (1 Li in 20 h) shows a sustained reversible electrochemical activity of 0.8  (e.g., 120 mAh/g), centered around 3.6 V vs
(e.g., 120 mAh/g), centered around 3.6 V vs  with limited polarization, as previously reported (Fig. 3). No efforts have been made to fine-tune the electrode formulation to near the theoretical 151 mAh/g; this constitutes the next stage of our research aimed at benchmarking samples made by solid-state, ionothermal, and polymer-based processes. Lastly, 12% loss in capacity (150 mAh/g for
with limited polarization, as previously reported (Fig. 3). No efforts have been made to fine-tune the electrode formulation to near the theoretical 151 mAh/g; this constitutes the next stage of our research aimed at benchmarking samples made by solid-state, ionothermal, and polymer-based processes. Lastly, 12% loss in capacity (150 mAh/g for  as compared to 170 mAh/g for
as compared to 170 mAh/g for  ) is compensated partially by a 4% gain in voltage (3.6 V instead of 3.45 V for
) is compensated partially by a 4% gain in voltage (3.6 V instead of 3.45 V for  . The main advantage, however, is the kinetics of the sulfate, being able to operate without carbon coating as opposed to
. The main advantage, however, is the kinetics of the sulfate, being able to operate without carbon coating as opposed to  . This results in a much better packing density, as "fast"
. This results in a much better packing density, as "fast"  is made of nanoparticles with very low tap density.
is made of nanoparticles with very low tap density.
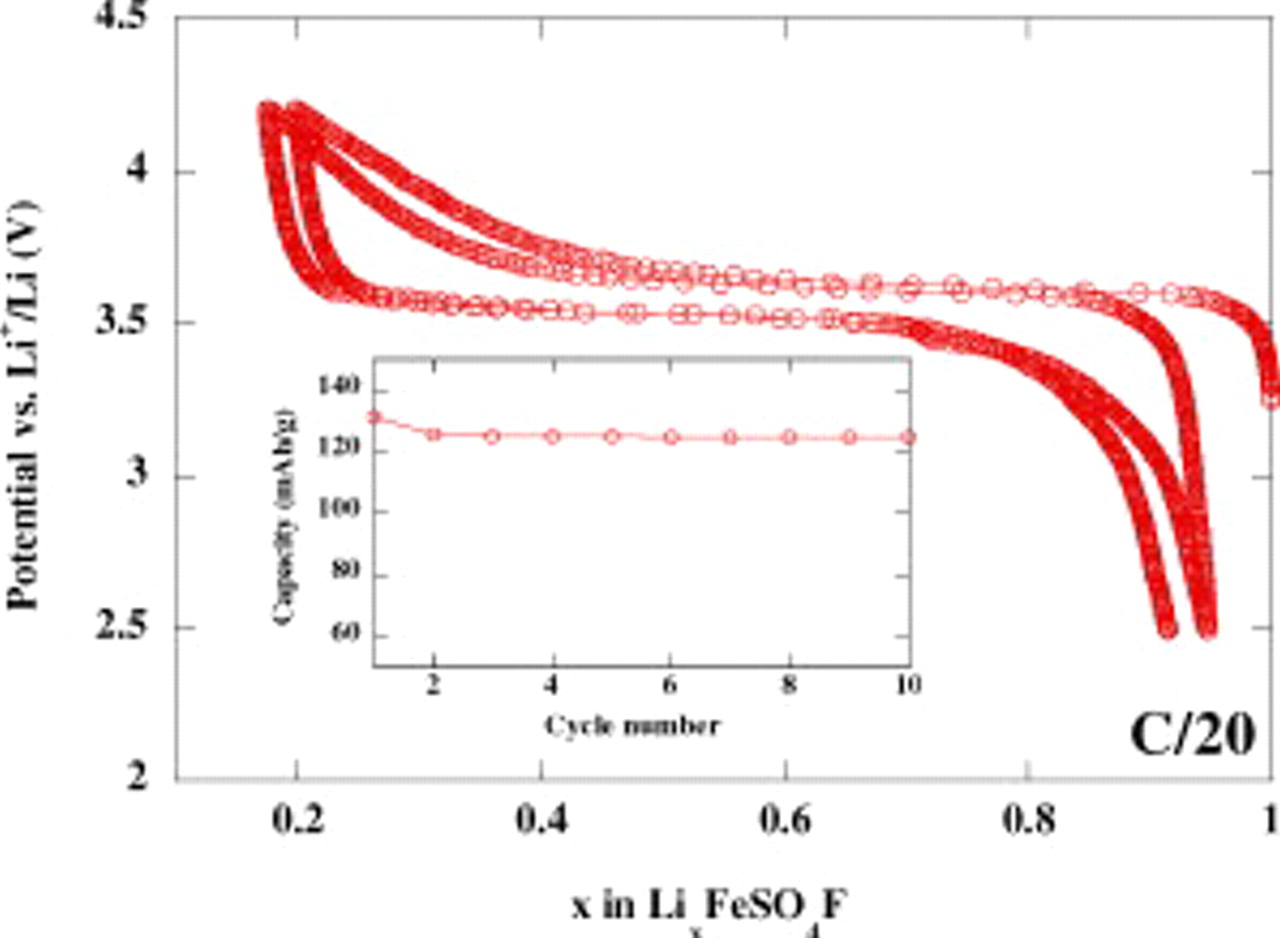
Figure 3. Electrochemical performances of the  material prepared with PEG
material prepared with PEG  polymer obtained by galvanostatic cycling of
polymer obtained by galvanostatic cycling of  of active material powder in Swagelok cell configuration using a rate of one lithium in 20 h (C/20) between 2.5 and 4.2 V vs Li. Inset: Its corresponding capacity retention over the first 10 cycles.
of active material powder in Swagelok cell configuration using a rate of one lithium in 20 h (C/20) between 2.5 and 4.2 V vs Li. Inset: Its corresponding capacity retention over the first 10 cycles.
Conclusions
We have presented a new synthetic approach for the low temperature preparation of  , which enlists the use of polymers. These polymers are solid at room temperature and liquid though melting or crossing
, which enlists the use of polymers. These polymers are solid at room temperature and liquid though melting or crossing  and thermally stable up to temperatures greater than
and thermally stable up to temperatures greater than  . The thermal stability of the polymers rather than their solvating properties is the key to a successful synthesis of
. The thermal stability of the polymers rather than their solvating properties is the key to a successful synthesis of  materials, which are electrochemically active with capacities approaching 121 mAh/g. Like ionic liquids, polymers do not work because of their solvating properties; their success is nested in their ability to modify the water release kinetics from the precursor
materials, which are electrochemically active with capacities approaching 121 mAh/g. Like ionic liquids, polymers do not work because of their solvating properties; their success is nested in their ability to modify the water release kinetics from the precursor  . However, the encapsulating efficiency of the polymers presented in this article is less than that of ionic liquids, which enable the synthesis of fluorosulfates in an open environment. Such an approach was implemented to the synthesis of other fluorosulfates containing either Na instead of Li or other 3d metals (Co or Ni instead of Fe). To provide a means to control the particle size and morphology using this new approach is the subject of an ongoing experimentation. In short, this approach provides new opportunities in the synthesis of fluorosulfates for practical applications as it bypasses both the cost associated with the use of ionic liquids and the lengthy reacting time linked to solid-state processes, while providing materials with identical performances. We hope it will lead to industrial processes.
. However, the encapsulating efficiency of the polymers presented in this article is less than that of ionic liquids, which enable the synthesis of fluorosulfates in an open environment. Such an approach was implemented to the synthesis of other fluorosulfates containing either Na instead of Li or other 3d metals (Co or Ni instead of Fe). To provide a means to control the particle size and morphology using this new approach is the subject of an ongoing experimentation. In short, this approach provides new opportunities in the synthesis of fluorosulfates for practical applications as it bypasses both the cost associated with the use of ionic liquids and the lengthy reacting time linked to solid-state processes, while providing materials with identical performances. We hope it will lead to industrial processes.
Acknowledgments
The authors grateful to C. Delacourt for helpful discussions regarding his expertise in solution chemistry and M. Courty for running the thermal analysis for the polymers that we have used for the present study.
Université de Picardie-Jules Verne assisted in meeting the publication costs of this article.